Das versteckte Erbe der Genregulation
Neue bioinformatische Methode identifiziert evolutionär konservierte Bereiche im Genom
Forschende des Max-Planck-Instituts für Molekulare Genetik und dem Berlin Institute of Health in der Charité (BIH) haben in einer Kooperation mit internationalen Kolleg*innen eine bioinformatische Methode entwickelt um evolutionär konservierte Bereiche im Genom zu identifizieren, die sich im Laufe der Zeit so stark verändert haben, dass ihre DNA-Sequenzen keine Übereinstimmung mehr aufweisen. Dr. Daniel Ibrahim ist Studienleiter und Letztautor der in Nature Genetics veröffentlichten Publikation.
, gesteuert durch evolutionär konservierte DNA-Sequenzen (rechts) von Maus (oben) und Huhn (unten), die keine Sequenzidentität mehr aufweisen")
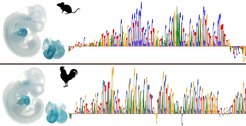
Bei der Entwicklung von Organen wie dem Herzen greifen evolutionär hochkonservierte genetische Programme, die bei Mensch und Tier in bemerkenswerter Weise übereinstimmen. So haben sich zum Beispiel die Aminosäuresequenzen der beteiligten Gene über hunderte Millionen Jahre nur wenig verändert.
Was sich aber stark verändert hat, sind die regulatorischen Bereiche im Genom, welche bestimmen, ob und wie diese Gene aktiv werden. Die Sequenzen dieser sogenannten cis-regulatorischen Elemente (CREs), auch Enhancer genannt, sind häufig nicht konserviert, insbesondere nicht zwischen evolutionär weit entfernten Spezies. Jedoch vermutet man, dass diese CREs trotz Sequenzunterschieden immer noch an derselben genomischen Position konserviert sein können, also funktionell konserviert sind. Das Ausmaß dieser Konservierung ist allderings unklar.
Genau dies wollten die Wissenschaftler*innen untersuchen. Dazu mussten sie herausfinden, welche Enhancer von derselben Vorgängersequenz abstammen. Sie analysierten regulatorische Genomabschnitte im Herz von Hühnern und Mäusen in unterschiedlichen embryonalen Entwicklungsstadien.
Zusammen mit der Forschungsgruppe von Professor Martin Vingron (MPIMG) und Boris Lenhard vom Imperial College in London entwickelten sie ein neues bioinformatisches Verfahren, mit dem man anhand der Position im Genom (also auf welchem Chromosom und wo dort genau) vorhersagen kann, wo die äquivalente Position in einem anderen Genom ist.
Der neu entwickelte Algorithmus identifizierte bis zu fünfmal mehr konservierte Enhancer als herkömmliche, auf Sequenzähnlichkeit basierende Methoden. Über epigenetische Markierungen, Machine-Learning Methoden und letztendlich im Mausmodell konnten die Forscher*innen zeigen, dass die Funktion der Enhancer trotz Sequenzunterschiede erhalten blieb. Die untersuchten konservierten regulatorischen Elemente hatten sich in ihrer Sequenz außeinanderetwickelt, aber dabei die Funktion beibehalten. Somit konnten die Wissenschaftler*innen beweisen, dass die Funktion der Enhancer konserviert sein kann, auch wenn sich die DNA-Sequenzen unterschieden.
“Ein zentrales Ergebnis unserer Arbeit ist ein bioinformatisches Werkzeug namens IPP (Interspecies Point Projection), das wir der wissenschaftlichen Community zur Verfügung stellen. Alle die interessiert daran sind zu wissen wo sich z.B. humane Enhancer im Genom der Maus oder Ratte oder Huhn oder Fisch befinden, können es sofort anwenden.”
Das neu entwickelte Verfahren kann bei der Diagnostik genetischer Erkrankungen beim Menschen helfen. Beispielsweise haben viele angeborene Herzfehler eine unklare genetische Ursache und sind möglicherweise auf Veränderungen in wichtigen Enhancern zurückzuführen.
So können mit dem Bioinformatik-Tool Erkenntnisse aus Tiermodellen auf den Menschen übertragen werden oder genetische Ursachen von Krankheiten im Tiermodell besser erforscht werden.
Link zur Publikation: Conservation of regulatory elements with highly diverged sequences across large evolutionary distances
News & Views: https://www.nature.com/articles/s41588-025-02194-2
Link zum IPP-Tool: https://github.com/tobiaszehnder/IPP
Link zum BIH: https://www.bihealth.org/de/aktuell/wenn-sich-die-dna-veraendert-aber-die-genetische-regulation-bleibt











