Gezielte Veränderung der DNA-Methylierung im Säugergenom
Forschungsbericht (importiert) 2018 - Max-Planck-Institut für molekulare Genetik
Die Rolle der DNA-Methylierung im Entwicklungs- und Krankheitsgeschehen
DNA-Methylierung bezeichnet eine chemische Veränderung der DNA, bei der Methylgruppen mithilfe von Enzymen, sogenannten Methyltransferasen, an einzelne Basen der DNA angehängt werden. Dies beeinflusst die Eigenschaften der DNA, ihre Sequenz wird dabei jedoch nicht verändert. Die Methylierung ermöglicht dem Organismus eine selektive Nutzung bestimmter DNA-Bereiche, indem durch diese einfache Modifikation zum Beispiel Gene gezielt stillgelegt werden können. DNA-Methylierung ist die zurzeit am besten untersuchte epigenetische Veränderung [1].
Schon vor etwa siebzig Jahren wurde die DNA-Methylierung im Säugergenom entdeckt, aber erst vor rund dreißig Jahren lernten wir aus genetischen Knockout-Studien, dass DNA-Methylierung beim Säuger für die normale Entwicklung unerlässlich ist, und in den letzten zehn Jahren konnten wichtige Einblicke in die Verteilungsmuster der Methylgruppen im Genom und deren dynamische Regulation im Verlauf der Entwicklung und bei der Entstehung von Krankheiten gewonnen werden [1, 2]. Heute finden Modulationen auf der Ebene des Epigenoms vor allem durch genetische oder pharmakologische Beeinflussung von regulatorischen Proteinen statt. Solche Experimente und die daraus gewonnenen Erkenntnisse sind sehr wertvoll, jedoch beeinflussen die veränderten Proteine das gesamte Genom und nicht nur gezielt eine Region oder einen spezifischen Genort, der aktuell für die Forschung von Interesse ist. Wir sind bislang noch nicht in der Lage, präzise bestimmte Regionen im Genom zu methylieren. Genau dies wäre jedoch erforderlich, um zu einem umfassenden Verständnis der Rolle der DNA-Methylierung bei der Regulation der Genaktivität zu gelangen.
Entwicklung von neuen Werkzeugen zur (Epi-)Genom-Editierung
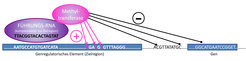
Mit der Entwicklung des CRISPR-Cas9-Systems sind die Möglichkeiten zur gezielten Editierung von Genomen sprunghaft gestiegen. Das zentrale Element dabei ist Cas9, eine Nuklease, die mithilfe von künstlichen „Führungs-RNAs“ (guide RNAs) gezielt an ausgewählte Regionen im Genom gelangt und dort die DNA schneiden kann. Dank der rasanten Entwicklung dieser Methode verfügen wir inzwischen auch über Cas9-Varianten ohne Nuklease-Aktivität. Diese sind genauso wie die aktive Variante in der Lage, mittels Führungs-RNAs gezielt bestimmte Regionen im Genom zu erreichen. Das Nuklease-inaktive Cas9 Protein kann flexibel an andere Effektorproteine, zum Beispiel Methyltransferasen, gekoppelt werden. Auf diese Weise hoffen wir, künftig gezielt einzelne Basen im Genom methylieren zu können (Abb. 1).
 ist an eine DNA-Methyltransferase (Dnmt3) gekoppelt. Dnmt3 wird dadurch gezielt in bestimmte Regionen im Genom geführt und kann dort seine Aktivität entfalten. Im Idealfall werden nur bestimmte Cytosine (C’s) methyliert, während die weiter entfernt liegenden Cytosine unverändert bleiben. Mit dieser Methode können wir genau erforschen, welche Methylierungen wann und wo für die Regulation des entsprechenden Gens verantwortlich sind.")
Dabei dürfen die folgenden Fragen nicht außer Acht gelassen werden:
* Welche Eigenschaften muss eine Region im Genom besitzen, damit sie methyliert werden kann?
* Kann jede einzelne Base/Position im Genom methyliert werden?
* Kann die Methylierung erfolgreich über längere Zeit aufrechterhalten werden?
* Bewirkt die Methylierung tatsächlich eine Stilllegung der zugehörigen Gene?
Unter weiterer Berücksichtigung von technischen und mechanistischen Aspekten haben wir nun ein System zur positionsspezifischen de-novo-Methylierung von DNA entwickelt. Um möglichst viele der daran beteiligten Einflussfaktoren wie beispielsweise endogene Enzyme, die Chromatin-Landschaft oder bereits vorhandene DNA-Methylierungen kontrollieren zu können und klare, interpretierbare Ergebnisse zu erhalten, verwenden wir eine Zelllinie, bei der die Methyltransferasen Dnmt3a/b komplett fehlen und die Funktion des für das Kopieren von DNA-Methylierungen wichtigsten Enzyms, der Methyltransferase Dnmt1, gezielt ein- und ausgeschaltet werden kann. Anhand dieses konditionellen Knockouts erhalten wir Zellen, die selbst keine neuen Methylgruppen ausbilden können. Sie sind jedoch in der Lage, vorhandene Methylierungsmuster zu kopieren. Um in diesen Zellen gezielt neue Methylgruppen einzuführen, die dann ebenfalls von Dnmt1 aufrechterhalten werden, haben wir, wie oben beschrieben, die Nuklease-inaktive Version von Cas9 mit einer DNA-Methyltransferase gekoppelt und können so die entstandenen Fusionsproteine in die Zellen einführen. Anschließend haben wir genomweit kontrolliert, an welchen Positionen neue Methylgruppen auftraten. So konnten wir sowohl die Zielgenauigkeit unseres Systems überprüfen als auch unerwünschte Methylierungen, sogenannte Off-Target-Effekte, feststellen [3].
Spuren des dCas9-Fusionssystems im Genom
Bei der Kontrolle der Methylierungsmuster in den Zellen nach Einführung des Fusionsproteins fanden wir zu unserer Überraschung eine universelle Aktivität, die unabhängig von der Anzahl der eingesetzten Führungs-RNAs und der Dauer der Induktion war. Wir stellten fest, dass jedes Cytosin im Genom mit Ausnahme von hoch geschützten CpG-reichen und konstitutiv unmethylierten Regionen methyliert wurde. Ein auf Cas9-basierendes Epigenom-Targeting scheint somit derzeit leider noch nicht geeignet, um gezielt nur definierte Positionen im Genom zu methylieren [3].
Auf dem Weg, aber noch nicht am Ziel
Die bisherigen Ergebnisse zeigen aber, dass eine gezielte Methylierung im Genom grundsätzlich möglich ist. Mit der bisher eingesetzten Methode werden allerdings momentan noch zu viele unerwünschte Positionen methyliert. Hinzu kommt, dass die neuen Methylierungsmuster nicht stabil waren; nach Ende der Induktion wurde die Methylierung wieder abgebaut. Wir sind aber davon überzeugt, dass diese Probleme überwunden und wir mit der Etablierung einer zielgenauen Methode zur de novo-Methylierung im Genom letztlich in der Lage sein werden, die genaue Rolle der DNA-Methylierung bei der Regulation der Transkription sowie deren Einfluss auf andere epigenetische Faktoren, wie zum Beispiel Histon-Modifikationen, aufklären zu können. Diese Erkenntnisse könnten dazu beitragen, neue epigenetische Methoden zur Behandlung von Krankheiten zu entwickeln, die durch Fehlexpression von Genen entstehen. Die jüngsten Errungenschaften im Bereich der Genom-Editierung ermöglichen die Durchführung von Experimenten, die bisher nicht einmal vorstellbar waren. Unser Labor ist führend in diesem Bereich und wir sind mit Begeisterung dabei, die neuen Technologien weiter voran zu treiben und auszubauen.
Literaturhinweise
Nature Reviews Genetics 14, 204–220 (2013)