Zwischen den Zeilen des Genoms lesen
Bislang unbekannter genetischer Mechanismus verursacht angeborene Fehlbildungen
Ein internationales Forschungsteam hat eine seltene genetische Erkrankung entdeckt, die sich in schweren Fehlbildungen der Gliedmaßen äußert. Wie die Forschenden in der Fachzeitschrift Nature beschreiben, liegt der Krankheit ein epigenetischer Mechanismus zugrunde, an dem Teile des Genoms mit bis dato unbekannter Funktion beteiligt sind. Der Prozess könnte auch die Ursache von anderen angeborenen Krankheiten sein.


Die regulatorischen Informationen in den nicht-kodierenden Anteilen des Genoms zu entschlüsseln – ist eine Odyssee auf der Suche nach genetischen Erkrankungen
Nachdem vor 20 Jahren das erste menschliche Genom sequenziert wurde, gab es zunächst eine große Überraschung. Man konnte lediglich 20.000 Gene identifizieren, die Baupläne für Proteine kodieren. Außerdem machten diese Gene weniger als zwei Prozent des Genoms aus, was die Frage nach der Funktion des viel größeren, nicht-kodierenden Teils aufwarf. Tatsächlich bezeichnete man die nicht-kodierende DNA ohne erkennbaren Nutzen zwischen den Genen lange als „Schrott“ (junk DNA). Heute ist klar, dass sich dort, zwischen den „Zeilen des Genoms“, wichtige Informationen befinden, um genetische Aktivität zur rechten Zeit am rechten Ort an- oder abschalten.
Ein internationales Forschungsteam aus Berlin und dem schweizerischen Lausanne entdeckte nun einen neuen Krankheitsmechanismus für eine genetische Erkrankung, verursacht durch genau solche nicht-kodierenden Sequenzen. Wie das Team im Fachjournal Nature erklärt, leistet das Ablesen eines DNA-Abschnitts in der Nähe des Entwicklungsgens engrailed-1 (En1) einen wesentlichen Beitrag für die Aktivierung dieses Gens. Das Gen En1 ist seit langem für seine zentralen Funktionen bei der Entwicklung der Extremitäten, des Gehirns, des Brustbeins und der Rippen bekannt. Die Wissenschaftlerinnen und Wissenschaftler berichten nun, wie die Aktivierung von En1 in den Gliedmaßen durch den neu identifizierten DNA-Abschnitt gesteuert wird und warum es bei einer Störung des Vorgangs zu schweren Fehlbildungen der Gliedmaßen kommt.
Ein Paradebeispiel für seltene Erkrankungen
„Ich erwarte, dass es noch mehr genetische Krankheiten mit vergleichbarer Ursache gibt, die sich nur bisher unserer Aufmerksamkeit entzogen haben“, sagt Stefan Mundlos, Forschungsgruppenleiter am Max-Planck-Institut für molekulare Genetik (MPIMG) in Berlin und Direktor des Instituts für Medizinische Genetik und Humangenetik an der Charité – Universitätsmedizin Berlin. „Die Ursachen für mehr die Hälfte der genetisch bedingten Krankheiten sind immer noch unbekannt, hier gibt es also noch viel Potenzial für weitere Forschung.“
Die Art der Fehlbildungen der drei in der Studie untersuchten Patienten ist außergewöhnlich. So sind die Knie beispielsweise nicht nach vorne gerichtet, einige Finger miteinander verschmolzen und es wachsen Nägel auf der Innenseite der Finger. „Offenbar ist während der Entwicklung der Gliedmaßen die Unterscheidung zwischen ventraler und dorsaler Seite – also der Handfläche beziehungsweise Fußsohle und der Rückseite – bei den Extremitäten verloren gegangen“, sagt Mundlos.
Die Patienten fielen zunächst Ärztinnen und Ärzten in Brasilien und Indien auf, die daraufhin DNA-Proben zur genetischen Untersuchung an Humangenetiker Andrea Superti-Furga an der Universität Lausanne schickten. Dessen Team entdeckte, dass bei allen Betroffenen ein ähnliches Stückchen nicht-kodierender DNA fehlte. Um der Sache auf den Grund zu gehen, taten sie sich mit Mundlos' Arbeitsgruppe in Berlin zusammen.
Fehlender DNA-Abschnitt sorgt für die Krankheit
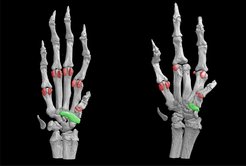
 und dem Krankheitsmodell einer Maus (rechts). Im Modell sind Fehlbildungen der Pfote sichtbar, wie verkleinerte (grün) oder fehlende (rot) ventrale Knochen.")
Am MPIMG machte sich die Wissenschaftlerin Lila Allou daran, die molekulare Ursache der Krankheit zu ergründen. „Anfangs wussten wir nur, dass bei den drei Patienten ein ähnliches kleines Stück Erbgut fehlte“, sagt sie. „Aber die Sequenz befand sich in einer großen genetischen Wüste – einem Abschnitt nicht-kodierender DNA, über den nichts wussten – weit entfernt von den nächsten zwei Genen.“
Der Nachweis, dass dieses fehlende Stück mit unklarer Funktion tatsächlich die Ursache für die Erkrankung war, gelang Allou mit Hilfe eines Mausmodells. Mit der CRISPR-Cas-Technologie entfernte sie die entsprechende DNA-Sequenz aus dem Mausgenom. „Mäuse mit der Deletion bildeten die Krankheit in großem Maße nach“, sagt Allou. „Die Ergebnisse bestätigten, dass der fehlende DNA-Abschnitt die Ursache der Erkrankung war.“
Weitere Untersuchungen von Mausembryonen zeigten dann, dass die genetisch veränderten Mäuse keine Aktivität des En1-Gens mehr in den Gliedmaßen aufwiesen, in ihnen das Gen also nicht angeschaltet wurde. En1 ist schon seit Jahrzehnten als besonders wichtiges Gen bekannt; seine Fehlregulation führte im vorliegenden Fall offenbar zu der Entwicklungsstörung. Auf welche Weise das fehlende Erbgutstückchen zum Verlust der En1-Aktivität führte, blieb jedoch vorerst noch im Dunkeln.
Dekodierung des nicht-kodierenden Bereichs
 und Knochen (rot) von Vorderpfoten einer normalen Maus (links) und dem Krankheitsmodell einer Maus (rechts). Im Modell sind Fehlbildungen wie zusammengewachsene Finger (Mitte), Polydaktylie (links) und ein ventral verschmolzener Finger (rechts) erkennbar.")

Die Forschenden stellten fest, dass ein RNA-Molekül in jener Region abgeschrieben wurde, die in den Betroffenen fehlte. Dieser nicht-kodierende DNA-Abschnitt stellte sich als des Rätsels Lösung heraus und wurde Maenli (für Master regulator of En1 in the Limb) getauft.
RNA fungiert meistens als Bote für Informationen und enthält den Bauplan für ein Eiweiß – doch in diesem Fall blieb die Information auf dem Molekül unübersetzt. „Diese Art von transkribierten Schnipseln findet sich in großer Zahl im Genom, welche davon wichtig sind und welche nicht, ist häufig schwer zu sagen“, sagt Allou. „Viele Wissenschaftler halten diese Moleküle für funktionslos, aber in diesem Fall hat es unser Interesse geweckt und wir wollten der Sache nachgehen.“
Die Wissenschaftlerin untersuchte die Funktion der Maenli-RNA, indem sie eine Mutation erzeugte, die die Transkription vorzeitig unterbrach. Mäuse mit einem inaktivierten Maenli zeigten dieselben Fehlbildungen wie die Tiere mit der Deletion – der Beleg, dass wirklich die fehlende RNA das Krankheitsbild in den Patienten hervorrief.
Außerdem schien es, als sei Aufbau und Sequenz des RNA-Moleküls nur zweitrangig. Wichtiger ist wohl die Aktivität selbst, also das Ablesen an dem jeweiligen Ort auf dem Erbgutstrang. Denn nachdem Allou die Sequenz durch einen völlig verschiedenen Abschnitt ersetzt hatte, zeigten die Mäuse zwar immer noch Anzeichen der Krankheit, aber weniger stark also durch die vollständige Inaktivierung von Maenli. Das Ablesen einer ganz anderen Sequenz an dieser Stelle reichte also offenbar aus, um das En1-Gen zu aktivieren – wenn auch in geringerem Maße als die ursprüngliche, natürliche Sequenz es vermochte.
Versteckte Überraschungen im Genom
Wie der abgelesene Schnipsel das En1-Gen genau aktiviert, ist noch unklar und Gegenstand der laufenden Forschung im Labor von Mundlos. Dennoch ist absehbar, dass die neuen Erkenntnisse weitreichende Folgen haben, sagt der Forscher: „Unsere Ergebnisse berühren die Fachgebiete der Humangenetik, RNA-Forschung, Genregulation und Entwicklungsbiologie“.
„Aus der Sicht der Entwicklungsbiologie haben wir einen neuen genetischen Mechanismus identifiziert, der während der frühen Embryonalentwicklung Zellen determiniert, zum ventralen Teil der Gliedmaßen zu werden“, ergänzt Allou. „Ich glaube, dass unsere Ergebnisse auch die zukünftige Diagnostik genetischer Erkrankungen beeinflussen wird und helfen kann, die Ursachen anderer seltener genetischer Krankheiten aufzuklären.“
„Über 90 Prozent der Genvarianten befinden sich im nicht-kodierenden Teil des Genoms, aber es ist sehr schwierig, sie zu deuten und für diagnostische Zwecke zu nutzen“, sagt die Wissenschaftlerin. „Unsere Arbeit führt uns klar vor Augen, dass genetische Varianten, die bisher ignoriert wurden, essentiell für das Verständnis der molekularen Ursachen von Krankheiten sein können.“ Um weitere solche ungeklärten Fälle zu lösen, müssten alle verfügbaren genetischen und epigenetischen Daten berücksichtigt werden – auch jene, die zuvor nicht so wichtig erschienen, sagt die Forscherin. „Wovon wir annehmen, dass es unwichtig ist, das könnte tatsächlich den Schlüssel für wesentliche Erkenntnisse bereithalten.“











