Better recognition of chromosomal diseases
Hi-C method improves detection of breaks and rearrangements in the genome
Breaks and rearrangements in the genome can lead to severe diseases, even if all genes remain intact. Hi-C, a method to map the three-dimensional structure of chromosomes, promises more reliable and accurate diagnoses of such defects, but is not used in the clinic yet. A team of researchers at the Max Planck Institute for Molecular Genetics and Charité – Universitätsmedizin Berlin led by human geneticists Malte Spielmann and Stefan Mundlos analyzed clinical samples from patients with genetic developmental disorders with the Hi-C method.

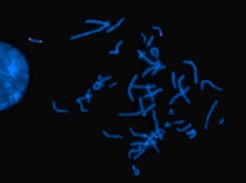
Chromosomes stained with fluorescence dyes under the microscope. While all DNA is stained blue, a specific sequence stained pink appears duplicated in one of the two copies of chromosome 17, but not the other. This part was duplicated and inserted in another part of the same chromosome, which leads to disease.
Already one mutation in the genome can have serious consequences. However, the loss of large sections of the genome or their relocation to new positions can also have dramatic effects. Often, cells with such defects are not even viable, since loss or change of genes leads to a loss of important functions.
Even if all genes remain intact after a chromosomal break, serious problems may occur. DNA segments responsible for the control of other genes can get to the wrong location, activating genes at the wrong time or place. Consequently, cancer, neurodegenerative diseases or developmental disorders may arise.
Examination of the genome’s loops
In spite of the vast progress in genetic testing, the identification of the genetic causes of such diseases remains difficult. “In about half of the cases a genetic diagnosis is not possible, leaving the patient with the uncertainty of the origin of the problem”, says Stefan Mundlos from the Max Planck Institute for Molecular Genetics and Charité – Universitätsmedizin Berlin. “In some cases, even sequencing the entire genome does not help."
As the team led by human geneticists Stefan Mundlos and Malte Spielmann describe in the current issue of The American Journal of Human Genetics, a method from basic research could improve clinical diagnostics considerably at some point in the future. The researchers applied a method called "Hi-C" (High-throughput Chromosome Conformation Capture) to samples from patients with developmental disorders suspected to be caused by chromosomal rearrangements. The Hi-C analysis shows, which parts of the genome come close to each other in the cell nucleus. Chromosomal rearrangements may alter these interaction patterns and can thus be seen in the analysis.
The team examined clinical samples from blood, skin and amniotic fluid of nine patients with chromosomal breaks but without damage to known genes. "We asked, can we use Hi-C to reproduce the clinical findings or can we even see more?", says Spielmann, who led the study together with Mundlos. “In fact, the results were much more complex than we expected.”
Hi-C untangles highly disorganized chromosomes
.")
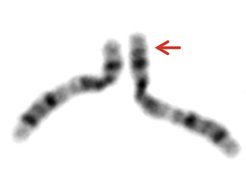
The classical analysis of chromosomal defects is done by a karyogram, which is a microscopic view of stained chromosomes. Another method, called comparative genome hybridization, works with fluorescent DNA snippets and shows gaps and duplications in the genome more precisely. However, both methods are relatively crude. “It is visible when something is wrong on a coarse scale, but it is difficult to say what exactly is wrong,” says Uirá Souto Melo. He, together with Rocio Acuna-Hidalgo and Robert Schöpflin, are the first authors of this publication.
It is important to not only see where the breaks in the genome are, but also which parts of the DNA molecule are in close contact with one another in the cell nucleus, the scientist explains. “DNA is not randomly packed in the nucleus”, explains Melo. “Instead, DNA is incredibly well-organized with multiple levels of organization and highly defined territories, although at first glance it doesn't look like that.” So far, the Hi-C method is the only way to accurately map the numerous slings and loops of DNA genome-wide.
Loops and triangles
Melo performed Hi-C on the patient’s cells by treating them with chemicals that permanently tether segments of neighboring DNA to each other first, then fragmenting the genome and finally sequencing the small pieces. Fragments that have originally been close in the cell nucleus occur together in the sequencing later.


After bioinformatic analysis, the frequency of contacts becomes visible in so-called heat maps, in which the color intensity of each point represents how often two genomic regions in the sample touched. “Parts of the genome with intense contacts within the region and some insulation towards the neighboring regions appear as characteristic triangular shapes in the heatmap”, says bioinformatician Robert Schöpflin. “Such regions form large loops within the DNA, which play an important role in functionally organizing regulatory sequences and genes.” The regions are called “topologically associated domains” (TADs); they represent regions of high interaction in the 3D space.
The importance of boundaries
Chromosomal breaks often lead to disruption of chromatin domains, sometimes with severe effects. “Imagine these domains as chambers in a glass tank – with oil, water and salt in the individual chambers,” says Melo. “When the boundary between them breaks, the contents mix and the composition in every chamber obviously changes.”
Similarly, without TAD boundaries, control functions from one domain spill over to another, as regulatory sequences from a DNA loop affect genes in a loop for which they are not responsible. This can lead to aberrant activation of genes by wrong regulators. “When a boundary that separates two domains is removed or if the content of two domains is swapped, a gene that is normally active in the developing limb may get activated, for example, in the brain”, says the scientist.
In the clinical samples examined, the team was able to not only confirm the existing findings and specify the effect of incorrectly organized TAD loops. They even found some additional breaks that had been overlooked by classical diagnostics.
The path to the clinic
“To be able to use Hi-C in the clinic tomorrow would be a dream,” says Melo. “But unfortunately, it is not possible yet.” The method is still too costly and complicated for routine tests. However, the scientist sees a lot of potential for optimization in the relatively new method. A lot of laboratory work could be automated, algorithms could be improved and it is possible to cut down on sequencing, says the researcher. “Right now, we have to reach out to our colleagues in human genetics and medicine all over the world in order to turn this laboratory technique into a real diagnostic method.”
[blk]











