3. Role of transposable elements in gene unit determination and evolution
From a cost-efficiency perspective, the human genome is an engineering nightmare. Since the completion of the human genome project in 2001, it has become clear that huge swaths of our genome are made of self-replicating genetic elements with the so-called “functional genome” haphazardly distributed in between them (Lander et al. 2001). Every single transposable element (TE) insertion to our genome is a massive burden: each element is copied trillions of times in a single organism (there are ~37 trillion cells in a human body). Moreover, TEs make up >90% of an average human gene and are degraded very quickly after they are transcribed. Why do we tolerate so many TEs in our (transcribed) genome? How did TEs reach such massive copy numbers? These are intensely studied questions with ever-surprising results. However, in practical terms, these elements become nucleation points for a plethora of nuclear RNA-binding proteins as soon as they are transcribed by RNAPII, which then affect the content and the fate of the resulting mRNA, even though these sequences themselves do not necessarily contribute to the translated part of our genome. Thus, any consistent gene-theory should take emergence, evolution, distribution, expression and suppression of transposable elements into account.
We will look deeply at ribonucleoprotein (RNP) composition and RNP formation immediately after, during and following transcription in order to build a comprehensive model that can be used to predict the amount and composition of an mRNA that will be produced from a given genomic locus. Furthermore, by integrating large amounts of biochemical data and evolutionary analysis, we would like to eventually understand the impact of anti-transposon/virus strategies on the way genes themselves evolve, from transcription start sites to the evolution of new exon-intron boundaries and termination sites, which altogether determine the complete mRNA molecule as an expression of its underlying gene.

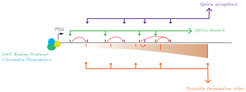
In the end, building such an accurate model can be used to design and evaluate artificial genetic circuits, to predict outcomes of clinically important mutations in RNA-binding proteins or RNA sequences, to develop possible solutions to reverse those outcomes, to treat genetic diseases and to understand how different cellular strategies against transposons/viruses might have shaped our genes and genomes over the course of our evolution.