Molekulare Ursachen genetisch bedingter kognitiver Störungen
Forschungsbericht (importiert) 2003 - Max-Planck-Institut für molekulare Genetik
Einführung
Zwei bis drei Prozent aller Menschen in unserer Bevölkerung haben einen Intelligenzquotienten (IQ) von <70 und gelten als geistig behindert. Unterhalb eines IQs von 50 spricht man von schwerer geistiger Behinderung; davon sind in Deutschland 300.000 bis 350.000 Menschen betroffen. Die Ursachen kognitiver Störungen sind noch großenteils unbekannt, jedoch sprechen epidemiologische Studien dafür, daß leichte kognitive Störungen (IQ 70-50) auf das Zusammenwirken einer Vielzahl von Genen und Umweltfaktoren zurückgehen und das untere Ende der normalen Intelligenzverteilung in der Bevölkerung darstellen. Demgegenüber scheinen schwere Formen der geistigen Behinderung ganz überwiegend auf definierte Chromosomenveränderungen (wie das häufige Down-Syndrom, verursacht durch ein überzähliges Chromosom 21) und Defekte einzelner Gene zurückzugehen. Dem gegenüber wurde die Bedeutung von Geburtskomplikationen und anderen exogenen prä- oder postnatalen Störungen als Ursache kognitiver Störungen in der Vergangenheit überschätzt.
Vermutlich können Defekte von weit mehr als 1.000 Genen Störungen der Hirnfunktion zur Folge haben. Jungen sind etwa 1,4-mal häufiger geistig behindert als Mädchen, was für eine besondere Rolle X-chromosomaler Gene spricht, jedoch findet sich der häufigste bekannte X-chromosomale Gendefekt, das Fragile X-Syndrom, nur bei zwei Prozent aller geistig behinderten Jungen. Bei ca. 5 Prozent aller Patienten hat man kleine Veränderungen im Bereich der Chromosomenenden als Krankheitsursache identifizieren können. Nach jüngsten Befunden ist es wahrscheinlich, dass derartige submikroskopische Veränderungen überall im menschlichen Genom vorkommen und für einen hohen Prozentsatz aller ungeklärten Fälle mit geistiger Behinderung verantwortlich sind.
Als eine der ersten Gruppen in Europa haben wir bereits vor 15 Jahren die neuen Möglichkeiten der Genomforschung genutzt, um die molekularen Ursachen von Erbkrankheiten aufzuklären. Dabei standen anfänglich erbliche Augenkrankheiten und Hörstörungen im Mittelpunkt [1]. Seit Mitte der neunziger Jahre haben wir uns zunehmend auf die Erforschung erblich bedingter kognitiver Störungen konzentriert, die in der klinischen Genetik, aber auch in der Krankenversorgung allgemein eine wichtige Rolle spielen. Nahezu die Hälfte aller genetischen Beratungen erfolgen zum Ausschluss erblich bedingter kognitiver Störungen, und auf die medizinische Versorgung und Betreuung geistig Behinderter entfallen ca. 8 Prozent der gesamten Aufwendungen im Bereich der Krankenversorgung [2]. Das Ziel unserer Untersuchungen ist die systematische Aufklärung der molekularen Ursachen ernster Formen der geistigen Behinderung und die Entwicklung einfacher diagnostischer Verfahren als Vorraussetzung für eine frühzeitige Diagnose und Prävention dieser Störungen in betroffenen Familien. Von der biochemischen und zellbiologischen Charakterisierung der betroffenen Gene versprechen wir uns außerdem neue Einblicke in die Pathogenese kognitiver Störungen und in die Entwicklung und Funktion des Gehirns.
Strategien für die molekulare Aufklärung von Gendefekten
Im Gegensatz zu anderen häufigen Krankheiten, die im Mittelpunkt der nationalen und internationalen Genomforschung stehen, gehen schwere kognitive Störungen großenteils auf Defekte einzelner Gene zurück. Der Zusammenhang zwischen diesen Krankheitsbildern und den beteiligten genetischen Faktoren ist daher sehr viel eindeutiger als bei Herz- und Kreislaufkrankheiten, Diabetes und Altersdemenz, die vom Zusammenwirken vieler Gene, aber auch von anderen, äußeren Faktoren wie der Lebensführung und vom Zufall abhängen.
Seit Beginn der Genomforschung konnten bereits in ca. 1500 verschiedenen Genen Defekte identifiziert werden, die beim Menschen erbliche Störungen mit einem einfachen, mendelschen Vererbungsmuster zur Folge haben. Die Sequenzierung des menschlichen Genoms hat die Aufklärung dieser ‚monogenen’ Krankheiten noch einmal entscheidend erleichtert. Dies gilt prinzipiell auch für die Suche nach den molekularen Ursachen schwerer kognitiver Defekte. Allerdings ist die Erkenntnis, dass die meisten dieser Störungen ebenfalls monogene Ursachen haben, offenbar noch nicht weit verbreitet. Dies ist wahrscheinlich einer der Gründe, weshalb ihre Erforschung noch ganz in den Anfängen steht. Andere Gründe sind das meist sporadische, also nicht familiäre Auftreten kognitiver Störungen, sowie die Tatsache, dass sich viele dieser Störungen klinisch nicht unterscheiden lassen (s.u.).
Im Rahmen unserer Untersuchungen zur Aufklärung der molekularen Grundlagen erblich bedingter kognitiver Defekte bedienen wir uns verschiedener, nachfolgend beschriebener Strategien, um die verantwortlichen Gene zunächst so genau wie möglich chromosomal zu lokalisieren.
Krankheitsassoziierte balancierte Chromosomenaberrationen: eine sichtbare Brücke zwischen Genotyp und Phänotyp
Bereits seit langem verfolgen wir das Konzept, Gendefekte durch Untersuchung von Patienten mit so genannten balancierten Chromosomenaberrationen zu identifizieren. Derartige Umlagerungen entstehen als Folge zweier Chromosomenbrüche mit anschließender Vertauschung der dadurch entsthenden Fragmente. Anhand der veränderten Chromosomenstruktur kann man sie unter dem Mikroskop meist einfach erkennen. Weil balancierte Chromosomenaberrationen nicht mit dem Verlust oder Zugewinn von Erbmaterial einhergehen, sind die meisten Träger solcher Veränderungen gesund. Etwa 6% aller reziproken Translokationen oder Inversionen führen jedoch zu klinischen Auffälligkeiten, die sich in vielen Fällen durch die Inaktivierung von Genen im Bereich der beteiligten Chromosomenbruchpunkte erklären lassen. Ca. die Hälfte dieser Patienten sind geistig behindert; daher ist die systematische molekulare Kartierung und Charakterisierung der Bruchpunktregionen bei derartigen Patienten eine viel versprechende Strategie für die Identifikation von Genen, die für die normale Hirnfunktion unentbehrlich sind.
Durch Rekrutierung von krankheitsassoziierten balancierten Chromosomenaberrationen (KBCA) im Rahmen eines weltweiten Netzwerks zytogenetischer Laboratorien und durch vielfältige andere nationale und internationale Kooperationen steht uns Untersuchungsmaterial von ca. 3.000 Patienten zur Verfügung. Zur Feinkartierung der betreffenden Chromosomenbruchpunkte bedienen wir uns der Fluoreszenz-In-situ-Hybridisierung (FISH, siehe Abb. 1).

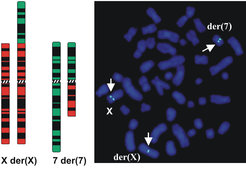
Hybridisierung einer X-chromosomalen DNS-Sonde auf Metaphase-Chromosomen einer mental retardierten Patientin mit einer balancierten Translokation der Chromosomen 7 und X ergibt Signale auf beiden rearrangierten Chromosomen und dem normalen X: die Sonde überspannt den Bruchpunkt.
Dabei werden klonierte, 30.000 bis ca. 1 Million Basenpaare lange menschliche DNS-Sonden mit Fluoreszenzfarbstoffen markiert und auf Chromosomen von Patienten mit KBCA hybridisiert. Dies führt zur Markierung der Abschnitte im Genom der Patienten, die den DNS-Sequenzen der Sonden homolog sind. Sonden, deren Sequenzen Bruchpunkte auf rearrangierten Chromosomen überspannen, kann man anhand der Hybridisierungsmuster einfach identifizieren.
Auf diese Weise haben wir in den vergangenen Jahren mehrere hundert chromosomale Bruchpunkte kartiert und kloniert. Bis heute konnten wir auf diese Weise mehr als 30 verschiedene Gene identifizieren, die bei Patienten mit kognitiven Defekten durch balancierte Chromosomenumlagerungen rearrangiert und inaktiviert waren. Die meisten dieser Gene sind im Hirn exprimiert, und für nicht wenige unter ihnen gibt es bereits unabhängige biochemische oder neurobiologische Hinweise auf eine bedeutende Rolle bei der Hirnentwicklung oder -funktion. Einige dieser Gene waren bei mehreren nicht verwandten Patienten mit KBCA betroffen, und die kausale Rolle mehrerer X-chromosomaler Gene konnten wir durch Nachweis von Mutationen in unabhängigen Familien mit X-chromosomal vererbter geistiger Behinderung zusätzlich belegen. Für eine Reihe weiterer Gene wird sich der Nachweis ihrer Bedeutung für die Entwicklung und Funktion des Gehirns nur experimentell erbringen lassen, etwa durch Erzeugung und Charakterisierung von Tiermodellen (V.Kalscheuer und Mitarbeiter; Kooperation mit N. Tommerup, Kopenhagen).
Matrix-CGH: eine hochauflösende Methode zur Erkennung submikroskopischer Chromosomenveränderungen
Eine andere erfolgreiche Strategie zur Feinkartierung von Gendefekten besteht in der Suche nach so genannten Mikrodeletionen oder -duplikationen, also submikroskopisch kleinen Veränderungen, die durch den Verlust oder Zugewinn von Chromosomenmaterial charakterisiert sind. Solche Mikrorearrangements sind als mögliche Ursache von Krankheiten mit ,monogenem’ Erbgang zwar schon seit langem bekannt, ebenso wie die Tatsache, dass Mikrodeletionen im Bereich der Chromosomenenden eine bedeutende Ursache für kognitive Störungen darstellen. Erst die Entwicklung der Matrix-CGH hat jedoch realistische Perspektiven für den Nachweis von Mikrodeletionen und -duplikationen im gesamten menschlichen Genom eröffnet.
Mit Unterstützung durch das BMBF haben wir uns bereits seit langer Zeit mit der Etablierung von Verfahren zum Nachweis submikroskopischer Imbalancen im menschlichen Genom befasst. Seit ca. zwei Jahren steht dabei die Optimierung und Einführung der Matrix-CGH im Mittelpunkt, deren Prinzip in Abbildung 2 dargestellt ist.
 oder dichten DNS-Sondenrastern (‚Matrix CGH’).")
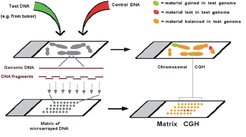
Diese Methode, eine Weiterentwicklung der etablierten ‚Comparativen Genomischen Hybridisierung’ (CGH), beruht auf der gleichzeitigen Hybridisierung unterschiedlich fluoreszenzmarkierter DNS von Patienten und gesunden Kontrollen auf ein dichtes Raster gleichmäßig über das Genom verteilter DNS-Sonden mit bekannter chromosomaler Lokalisation. Gegenüber der konventionellen CGH hat die Matrix-CGH eine wesentlich höhere Auflösung; bei Verwendung eines in unserer Abteilung hergestellten Rasters aus 4.250 gleichmäßig über das Genom verteilten Sonden kann man die meisten Mikrodeletionen mit einer Größe von 1 Million Basenpaaren sicher erkennen.
Serielle Analysen an gesunden Probanden und zytogenetisch voruntersuchten, geistig behinderten Patienten mit klinischem Verdacht auf Chromosomenaberrationen haben gezeigt, dass sich diese Methode zur Diagnose submikroskopischer unbalancierter Chromosomenveränderungen eignet und für diese Fragestellung sowohl der klassischen Karyotypanalyse als auch der konventionellen CGH überlegen ist (Abb. 3).


DNS von Patienten und gesunden Kontrollen wurde mit verschiedenen Fluoreszenzfarbstoffen markiert und gleichzeitig auf ein Raster aus 4.250 DNS-Sonden hybridisiert, deren relative Signalintensitäten bei Patienten und Kontrollen hier dargestellt sind. Jeder Punkt entspricht einer DNS-Sonde; links: Chromosom 1, 2 usw.; rechts: Sonden des X- und Y-Chromosoms.
Durch Erhöhung der Sondendichte könnte man die Auflösung dieser Methode noch deutlich weiter steigern, was die sichere Erkennung sehr viel kleinerer genomischer Imbalancen erlauben würde. Als bisher einzige Gruppe in Deutschland, aber parallel zu ähnlichen Bemühungen von Gruppen in Kanada, den USA, England und den Niederlanden haben wir uns daher entschlossen, für alle menschlichen Chromosomen Raster aus überlappenden DNS-Sonden herzustellen, mit dem Ziel, das gesamte menschliche Genom lückenlos abzudecken. Gegenüber den heute verwendeten CGH-Rastern wird dies die Auflösung dieser Methode nochmals um den Faktor 10 verbessern. Zur Herstellung dieser Sonden verwenden wir DNS von 32.450 charakterisierten und Sequenz-verifizierten BAC-Klonen, die im Mittel 200.000 Basenpaare lange menschliche DNS-Fragmente enthalten (Kooperation mit P. de Jong, Oakland). In den vergangenen Monaten ist es uns bereits gelungen, überlappende Sondenraster für die Chromosomen X, 4 und 22 herzustellen, und bei ersten Untersuchungen mit diesen DNS-Chips haben wir neben einer beträchtlichen Zahl ‚normaler’ Gendosisvarianten bereits bei mehreren Patienten spezifische, submikroskopische Chromosomenveränderungen identifiziert.
In enger Abstimmung mit anderen auf diesem Gebiet tätigen Gruppen wird es jetzt zunächst darum gehen, diese auffälligen Befunde mithilfe etablierter Verfahren (wie der FISH) zu überprüfen und Zug um Zug hochdichte Raster für alle menschlichen Chromosomen herzustellen. Parallel dazu haben wir begonnen, in Familien mit X-chromosomal vererbter geistiger Behinderung systematisch nach submikroskopischen Veränderungen des X-Chromosoms zu suchen. Zur Häufigkeit von Imbalancen im gesamten Genom von Patienten mit kognitiven Störungen gibt es bisher keine verlässlichen Daten; aufgrund der Häufigkeit von Mikrodeletionen im Bereich der Chromosomenenden dürften sie jedoch eine bedeutende Rolle spielen, für die Diagnostik und als Ausgangspunkt für die Identifikation der beteiligten Gene.
X-chromosomale Formen der geistigen Behinderung: Systematische Suche nach Mutationen in Kandidatengenen
Die Tatsache, dass deutlich mehr Jungen als Mädchen geistig behindert sind, ist schon vor vielen Jahrzehnten als Hinweis auf eine wichtige Rolle des X-Chromosoms gedeutet worden, da die Auswirkungen von Defekten X-chromosomaler Gene bei Mädchen durch das Vorhandensein eines zweiten, normalen X-Chromosoms abgeschwächt werden können, bei Jungen jedoch nicht. Seit Beginn der 90er-Jahre haben wir uns zunehmend mit dieser Frage auseinandergesetzt und mit der Gründung des ‚European X-linked Mental Retardation Consortiums’ (EURO-MRX; Chelly et al, Paris; Frijns et al, Leuven; Hamel et al, Nijmegen; Moraine et al, Tours; Ropers et al, Berlin) die organisatorischen und logistischen Voraussetzungen für eine systematische Erforschung nicht syndromaler Formen der X-chromosomalen mentalen Retardierung (NS-XLMR) geschaffen. Inzwischen verfügen wir über Zellinien und DNS von mehr als 450 nicht verwandten Familien mit X-chromosomaler geistiger Behinderung, die weltweit größte Sammlung dieser Art. Durch Analyse der Kopplungsintervalle von 125 Familien mit NS-XLMR gelang es uns zu ermitteln, wie sich die relevanten Gendefekte auf dem X-Chromosom verteilen. Diese Untersuchungen ergaben, dass ca. ein Drittel aller verantwortlichen Mutationen zentromernah auf dem kurzen Arm des X-Chromosoms lokalisiert sind [3], wie in Abbildung 4 dargestellt.
.")

Durch systematische Mutationssuche in Familien mit überlappenden Kopplungsintervallen konnten wir in dieser Region bisher 4 neue Gene für NS-XLMR identifizieren, und auch an der Aufklärung der meisten anderen bis heute bekannten Gene hatten wir und andere Mitglieder des EURO-MRX-Konsortiums maßgeblichen Anteil. Jedes einzelne der bisher identifizierten Gene ist jedoch nur in einem sehr geringen Prozentsatz aller untersuchten Familien mit NS-XLMR mutiert. Dies spricht dafür, dass es auf dem menschlichen X-Chromosom zwischen 50 und 100 Gene für non-syndromale geistige Behinderung gibt, und dass erst 20 bis 40 Prozent dieser Gene bekannt sind.
Das Ziel unserer Untersuchungen ist die Identifikation möglichst vieler X-chromosomaler Gendefekte, die zu ‚unspezifischen’ oder syndromalen Formen der XLMR führen, die Aufklärung der Funktion und Interaktion dieser Gene bei Gesunden und Patienten sowie die Entwicklung einfacher molekular-diagnostischer Methoden für den Nachweis dieser häufigen Störungen.
Homozygotie-Kartierung in großen blutsverwandten Familien: ein Königsweg zur Identifikation häufiger rezessiver Gendefekte
Mindestens 40 Prozent aller schwer geistig behinderten Patienten in unserer Bevölkerung sind sporadisch auftretende, sogenannte ‚idiopathische’ Fälle mit unbekannter Krankheitsursache. Es spricht vieles dafür, daß den meisten dieser sporadischen Fälle dominante oder rezessive Defekte autosomaler (also nicht X- oder Y-chromosomaler) Gene zugrunde liegen, da Träger schwerer dominanter Störungen selbst betroffen sind und deshalb meist keine Kinder haben, rezessive Störungen in Kleinfamilien jedoch sehr selten mehr als einmal auftreten, auch wenn die Krankheit in der Bevölkerung recht häufig ist. Die Aufklärung der molekularen Ursachen autosomal rezessiver Formen der geistigen Behinderung ist außerdem durch die Tatsache erschwert, daß sich viele Formen klinisch nicht unterscheiden lassen, was derartige Untersuchungen in unserer Bevölkerung praktisch unmöglich macht.
Aus diesem Grunde haben wir mit Forschungsinstituten im Iran und in Indien Kooperationsabkommen zur Aufklärung autosomal rezessiv vererbter kognitiver Defekte geschlossen. Iranische und indische Familien sind im Allgemeinen bedeutend größer als deutsche Familien, was sich auch in der Altersverteilung ausdrückt: so ist im Iran 70 Prozent der Bevölkerung jünger als 30 Jahre. Für die geplanten Untersuchungen noch wichtiger ist jedoch die extrem große Häufigkeit von Verwandtenehen: in mehreren iranischen Subpopulationen haben 60 Prozent der Kinder blutsverwandte Eltern, und die Verhältnisse sind in weiten Teilen Indiens ähnlich. In beiden Ländern sind Familien mit mehreren geistig Behinderten ausgesprochen häufig, was die wichtige Rolle autosomal rezessiver Gendefekte in der Ätiologie schwerer kognitiver Störungen belegt.
Blutsverwandte Eltern haben häufiger Kinder mit autosomal rezessiven Erbkrankheiten, da bei ihnen des Risiko, dieselbe defekte Erbanlage zu tragen und weiterzugeben wie ihr Ehepartner, sehr viel größer ist als bei nicht verwandten Eltern. Nun vererben sich diese Erbanlagen nicht unabhängig voneinander, sondern gemeinsam mit benachbarten Genen als Teil eines zusammenhängenden Chromosomenabschnitts. Kranke Kinder aus solchen Verwandtenehen erben deshalb von ihren Eltern nicht nur zwei identische Kopien desselben defekten Gens: auch die flankierenden Abschnitte auf beiden Chromosomen sind völlig gleich. Normalerweise unterscheiden sich homologe Chromosomenabschnitte in einer Vielzahl von DNS-Bausteinen, sogenannten ‚single nucleotide polymorphisms’ oder ‚SNPs’, die man mit verschiedenen einfachen Methoden nachweisen kann. Das Fehlen jeglicher genetischer Unterschiede zwischen zwei homologen Chromosomenabschnitten von Patienten ist daher sehr auffällig und läßt sich bei Kindern blutsverwandter Eltern zur Kartierung autosomal rezessiv vererbter Gendefekte nutzen.
Wir haben bislang 9 konsanguine Familien mit mehreren schwer geistig Behinderten im Iran rekrutiert und in der DNS dieser Patienten mithilfe von DNA-Chips nach homozygoten Chromosomenabschnitten gesucht (Kooperation mit F. Rüschendorf und P.Nürnberg, MDC Berlin). Die bisherigen Ergebnisse dieser Studien sind ermutigend: bei allen bisher untersuchten Familien konnten wir umschriebene homozygote Genomabschnitte identifizieren, und in zwei nicht verwandten Familien aus verschiedenen Gegenden Irans fanden wir nahezu identische, weitgehend überlappende homozygote Abschnitte. Diese Befunde könnten dafür sprechen, daß sich hier ein relativ häufiger Gendefekt verbirgt, analog zu ähnlichen Befunden bei rezessiven Formen der Taubheit, die zu nahezu 50 Prozent auf das Konto von Mutationen in einem einzigen Gen gehen [4].
Das Nahziel dieser Untersuchungen ist die molekulare Aufklärung der kognitiven Defekte in diesen 9 Familien. Aufgrund unserer Kooperationsvereinbarungen mit Partnern in Indien und vor allem im Iran, wo bereits ein landesweites Netzwerk von 400 genetischen Beratern existiert, haben wir Zugang zu einer praktisch unlimitierten Zahl konsanguiner Familien. Nach dem ermutigenden Ergebnis unserer Voruntersuchungen beabsichtigen wir daher, diese Studien auf größere Kohorten blutsverwandter Patienten und Familien auszuweiten, sofern es uns gelingt, dafür finanzielle Unterstützung zu bekommen. Dabei denken wir an die Untersuchung von 250 Familien innerhalb von 5 Jahren, mit dem Fernziel, die molekularen Ursachen aller häufigen autosomal rezessiven Formen der geistigen Behinderung zu identifizieren (H.H.Ropers, S.Lenzner und Mitarbeiter). Bisher ist erst ein einziges Gen für autosomal rezessive geistige Behinderung bekannt [5].
Zusammenfassung und Ausblick
Durch systematische Untersuchungen an geistig behinderten Patienten mit balancierten Chromosomenaberrationen und durch gezielte Mutationssuche in Familien mit X-chromosomalem Erbgang haben wir bis heute über 40 verschiedene Gendefekte identifiziert, die beim Menschen mit kognitiven Störungen einher gehen. Ihr funktionelles Spektrum ist breit: mehrere dieser Gene sind an der Steuerung des Wachstums von Nervenzellfortsätzen beteiligt, andere an der Transmission elektrischer Signale in Synapsen, den Schaltstellen von Nervenzellen; wieder andere Gene haben eine Rolle bei der Genregulation, der Modifikation von DNS, RNS oder Proteinen oder beim Abbau von Eiweißen. Für viele dieser Gene sind jedoch weder die eigentlichen Reaktionspartner in der Zelle noch die Mechanismen bekannt, welche bei Patienten zur geistigen Behinderung führen.
Die Aufklärung der biochemischen, zellbiologischen und neuroanatomischen Veränderungen, welche diesen Störungen zugrunde liegen, wird uns daher noch lange beschäftigen (S. Schweiger, C.Scharff, U. Nuber, D.Walther und Mitarbeiter, s. http://www.molgen.mpg.de/research/ropers/) und in vielen Fällen die Erzeugung von Tiermodellen erforderlich machen (Kooperation mit S. Sigrist, Göttingen). Ein besonders interessanter Aspekt dieser Untersuchungen ist die Frage, ob und in wieweit Varianten dieser Gene auch für die Variabilität der Intelligenz in der normalen Bevölkerung verantwortlich sind, ob es sich dabei also um echte ‚Kognitionsgene’ handelt. In diesem Zusammenhang ist es bemerkenswert, daß das von uns kürzlich identifizierten ZNF41-Gen, welches für einen X-chromosomalen Transkriptionsfaktor kodiert, im Genom der Maus nicht vorkommt [6].
Einem Großteil der sporadischen Fälle mit ‚idiopathischer’ geistiger Behinderung liegen wahrscheinlich autosomal rezessive Gendefekte oder submikroskopische Chromosomenveränderungen zugrunde. Die Ausweitung unserer Untersuchungen auf autosomal rezessive Formen der geistigen Behinderung und auf kleine genomische Imbalancen dürfte daher nicht nur für das Verständnis der normalen und gestörten Hirnfunktion, sondern auch für die Diagnostik und Prävention kognitiver Störungen weitreichende Konsequenzen haben.