Untersuchung von Bindungsstellen zur Aktivierung von Genen
Forschungsbericht (importiert) 2004 - Max-Planck-Institut für molekulare Genetik
Nach der Sequenzierung des Humangenoms und zahlreicher weiterer Genome konzentrieren sich heute wesentliche Forschungsanstrengungen auf die Analyse der gewonnenen Daten. Diese beschränkt sich jedoch nicht auf die reine Annotation der Gene, d.h., ihre Identifizierung und Zuordnung zu bestimmten Funktionen. Die Kenntnis der gesamten DNA-Sequenz eines Organismus erlaubt vielmehr eine Reihe von neuen Fragen, insbesondere nach den Mechanismen der Regulation von Genen. Heute interessiert uns nicht nur, welche Funktion ein Gen bzw. das von ihm kodierte Protein besitzt, sondern auch, welche Mechanismen dazu führen, dass das Gen überhaupt aktiviert, also abgelesen und in ein Protein übersetzt wird.
Transkriptionsfaktoren als Regulatoren der Genexpression

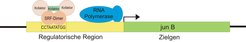
Der menschliche Organismus besitzt ca. 25.000 Gene, die während seines gesamten Lebens in jeder seiner Zellen vorhanden sind. In jeder Phase der Entwicklung und in jeder Zellart werden aber unterschiedliche Teilmengen dieses Genpools aktiviert. Heute kennen wir zumindest teilweise die biologischen Mechanismen, die für diese Aktivierung verantwortlich sind. Von besonderer Bedeutung ist eine Gruppe von DNA-bindenden Proteinen, die so genannten Transkriptionsfaktoren. Sie bilden einen Komplex mit dem Enzym RNA-Polymerase, das für das Ablesen der DNA verantwortlich ist und aktivieren es dadurch. Die Transkriptionsfaktoren erkennen bestimmte Sequenzmuster, die am Startpunkt eines Gens auf der DNA angeordnet sind (Abb. 1). Das Interesse der Abteilung Bioinformatik am Max-Planck-Institut für molekulare Genetik konzentriert sich auf die Identifikation solcher Sequenzmuster und die Frage, in welcher Kombination Transkriptionsfaktoren über Bindung an diese Muster bestimmte Gene aktivieren.

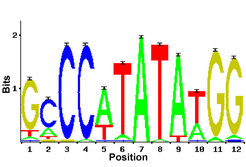
Molekularbiologen und Biochemiker beschäftigen sich schon seit Jahren mit Sequenzmustern, an welche Transkriptionsfaktoren binden können. Inzwischen wissen wir, dass viele Transkriptionsfaktoren an jeweils mehrere unterschiedliche Muster im Genom binden. Der Transkriptionsfaktor SRF (serum response factor) beispielsweise bindet an die Basenfolge CCTAATATGG vor dem Gen junB und trägt dadurch zu dessen Aktivierung bei (Abb.1). SRF bindet aber auch an anderen Stellen im Genom, die sich von der o.g. Sequenz in verschiedenen Positionen unterscheiden. Seine Bindungsstellen werden daher allgemein durch die Abfolge der möglichen Basen oder Basenalternativen beschrieben. Abstrakter ist die Beschreibung einer Bindungsstelle durch so genannte „Gewichtsmatrizen“. Diese geben für jede Position einer zu beschreibenden Bindungsstelle die mögliche Verteilung der Basen an (Abb. 2).
Phylogenetic footprinting - Identifikation wichtiger biologischer Signale durch evolutionäre Konservierung
Ein grundsätzliches Problem bei der Analyse von Bindungsstellen ist die Tatsache, dass eine definierte, kurze Abfolge von Basen innerhalb des Gesamtgenoms sehr oft vorkommt. Die Beschreibung der Bindungsstelle als Abfolge von Basen reicht daher nicht aus, um vorherzusagen, an welcher Stelle im Genom ein Transkriptionsfaktor tatsächlich bindet. Wichtige regulatorische Sequenzen sind allerdings häufig evolutionär konserviert. Bioinformatiker versuchen daher, durch den Vergleich der Genomsequenzen verschiedener Organismen Hinweise auf die Bedeutung einer bestimmten Sequenz (= potenziellen Bindungsstelle) zu erhalten. Umgekehrt stellen konservierte Sequenzen innerhalb regulatorischer Regionen primäre Kandidaten für die Suche nach neuen Bindungsstellen für Transkriptionsfaktoren dar; entsprechend werden sie intensiv in Hinblick auf Übereinstimmungen mit bekannten Bindungsmustern untersucht. Die Sequenz der bereits erwähnten Bindungsstelle für SRF wäre wenig informativ, wenn sie allein im Humangenom betrachtet wird. Die gleiche Sequenz tritt interessanterweise in vergleichbarer Position vor einem Gen auch im Genom der Maus auf. Der Vergleich beider Genome ergibt so einen starken Hinweis darauf, dass es sich bei diesem Muster um ein wichtiges biologisches Signal handeln könnte. Diese Herangehensweise wird als phylogenetic footprinting bezeichnet. Die Wissenschaftler der Abteilung Bioinformatik am MPI für molekulare Genetik haben eine Reihe von Computerprogrammen entwickelt, um solche konservierten Bereiche vor orthologen Genen von Mensch und Maus zu identifizieren und anschließend mit bereits bekannten Bindungsstellenmustern zu annotieren. Die Informationen sind in der Datenbank CORG (Comparative Regulatory Genomics) gespeichert und unter http://corg.molgen.mpg.de/ öffentlich zugänglich.
Identifikation der Zielgene von Transkriptionsfaktoren
Die Vorhersage von evolutionär konservierten Bindestellen kann auch helfen, Zielgene von Transkriptionsfaktoren zu identifizieren. In einer Zusammenarbeit mit A. Nordheim von der Universität Tübingen wurden zunächst mittels DNA-Chip-Experimenten (siehe unten) die putativen Zielgene von SRF ermittelt. Unter diesen befanden sich sowohl solche Gene, die von SRF direkt reguliert wurden, als auch diejenigen Gene, die erst in Folge der Aktivierung durch SRF aktiviert wurden. Aus dieser Gesamtmenge konnten mithilfe der beschriebenen Methoden der Mustersuche und der Analyse der evolutionären Konservierung diejenigen Gene herausgefiltert werden, die direkt von SRF beeinflusst werden. Diese Information war hilfreich, um einen bislang unbekannten Mechanismus der Differenzierung von Muskelzellen aufzuklären [1].
Analyse von Aktivierungsmustern


Für komplexe biologische Abläufe ist aber nicht nur die Aktivierung von Einzelgenen, sondern insbesondere die aufeinander abgestimmte Aktivierung ganzer Gruppen von Genen von Bedeutung. Solche „Aktivierungsmuster“ können mithilfe von DNA-Chips bestimmt werden (siehe von Heydebreck et al., Genexpressionsanalyse komplexer klinischer Phänotypen mittels DNS-Arrays, MPG-Jahrbuch 2003). Aus diesen Experimenten kann man ablesen, welche Gene sich in definierten Zellarten unter definierten Bedingungen ähnlich verhalten; sie werden als ko-exprimierte Cluster von Genen bezeichnet. Die Arbeitsgruppe beschäftigt sich unter anderem mit der Frage, ob eine solche Koexpression auf gemeinsame regulatorische Mechanismen, beispielsweise gleiche Transkriptionsfaktoren, zurückzuführen ist. Mithilfe der CORG-Datenbank untersuchen die Wissenschaftler die regulatorischen Bereiche der koexprimierten Gene. In erster Näherung katalogisieren sie die evolutionär konservierten Bindestellen innerhalb dieser Bereiche, weil diese auf eine regulatorische Funktion des jeweils bindenden Faktors hinweisen. Die evolutionär konservierten Bindungsstellen innerhalb der regulatorischen Bereiche sind aber immer noch zu zahlreich, als dass sie bereits Aufschluss über eine bestimmte Funktion geben könnten. Daher vergleichen die Wissenschaftler die Häufigkeit des Auftretens einer gewissen Bindestelle innerhalb eines koexprimierten Clusters mit der Wahrscheinlichkeit ihres zufälligen Auftretens. Eine solche Analyse wurde beispielsweise anhand von öffentlich verfügbaren DNA-Chip-Daten zum Zellzyklus in einer menschlichen Zelllinie durchgeführt. Die Koexpressionscluster bestanden aus den Genen, die jeweils in einer bestimmten Phase des Zellzyklus stark exprimiert sind. Die Analyse ihrer regulatorischen Muster ergab eine Reihe von Transkriptionsfaktoren, die nach der beschriebenen statistischen Schlussweise eine wichtige Rolle für den Zellzyklus spielen (Abb. 3). Ein Vergleich mit experimentellen Ergebnissen bestätigte dies [2].
Interaktion von Transkriptionsfaktoren untereinander
Zurzeit arbeiten die Wissenschaftler der Abteilung Bioinformatik an der Entwicklung von Methoden, um das Zusammenspiel der Transkriptionsfaktoren zu studieren. In dem im Vergleich zum Säugerorganismus einfacheren Fall der Genregulation in Hefe konnten sie zeigen, dass Transkriptionsfaktoren, deren Bindungsstellen häufig in räumlicher Nähe zueinander auf einer Sequenz zu finden sind, oft auch physikalisch miteinander in Interaktion treten [3]. Auf der Suche nach ähnlichen Prinzipien bei Säugetieren suchen sie nun nach statistischen Tendenzen in der Kombination von Bindungsstellen auf der DNA. Hier kommt den Forschern erneut die evolutionäre Konservierung zu Hilfe und ermöglicht eine Reduktion der falsch positiven Vorhersagen. In einer auf diese Art reduzierten Liste von Transkriptionsfaktor-Paaren, deren Bindestellen häufig nahe beisammen auftreten, werden tatsächlich viele bekannte, miteinander interagierende Faktoren gefunden [4].
Die beschriebenen Arbeiten beruhen auf einer detaillierten Aufarbeitung und mathematischen Durchdringung der vorhandenen, experimentell ermittelten Daten. Bekannte Bindungsstellen von Transkriptionsfaktoren müssen miteinander verglichen und gruppiert werden, um Doppelzählungen zu vermeiden. Ihr Informationsgehalt wird berechnet und innerhalb der Arbeitsgruppe wurden neue mathematische Verfahren entwickelt, um die zu erwartende Anzahl der falsch positiven Funde – mithin die statistische Signifikanz - vorherzusagen [5]. Ein häufig auftretendes Problem bei der Analyse von Daten aus der funktionalen Genomforschung ist das multiple Testen: Aufgrund der großen Menge an Daten kann eine Hypothese mit Hilfe vieler Fälle getestet werden. Dies führt aber nur scheinbar zu beeindruckenden Signifikanzwerten. Um eine realistische Aussage zu erhalten, müssen die resultierenden Signifikanzwerte entsprechend der Anzahl der durchgeführten Tests korrigiert werden, um eine adäquate Auswertung von biologischen Daten zu gewährleisten.
Die Regulation der Ausprägung eines Gens wird nicht nur von Transkriptionsfaktoren bestimmt. Trotzdem muss diese Ebene der Regulation studiert werden, um ein Gesamtbild der verschiedenen regulatorischen Mechanismen einer Zelle aufzeigen zu können. Damit kommen wir dem umfassenden Verständnis der Funktion einer lebenden Zelle einen wesentlichen Schritt näher.