Molekulare Mechanismen der Skelettentwicklung
Forschungsbericht (importiert) 2005 - Max-Planck-Institut für molekulare Genetik
Einleitung
Angeborene Fehlbildungen des Skeletts treten relativ häufig auf, besonders oft sind die Extremitäten betroffen. Diese Gruppe von Erkrankungen kann in verschiedene, klinisch relevante Untereinheiten eingeteilt werden, die intensiv erforscht werden, um ihre Ursachen aufzudecken. Grob einordnen lassen sie sich in
- Polydaktylien (die Vermehrung von Fingern),
- Reduktionsanomalien (den Verlust von einzelnen Elementen),
- Brachydaktylien (die Verkürzung von einzelnen Elementen),
- Syndaktylien (die Verschmelzung einzelner Finger).
Die genetischen Ursachen für diese Vielzahl von Phänotypen sind vielfältig [1, 2]. Wie bei kaum einem anderen Organ treffen bei der Entwicklung des Skelettsystems komplexe Mechanismen der Musterbildung, der Organogenese und des Wachstums aufeinander, die durch eine Vielzahl von Signalsystemen gesteuert werden. Die Erforschung dieser komplexen Signalregulation ist Schwerpunkt der Arbeit der Forschungsgruppe Entwicklung & Krankheit.
Die Bildung von Gelenken und ihre Defekte
Die frühe knorpelige Anlage der Extremitätenknochen wird zunächst als kontinuierliches Element angelegt, das Ellenbogengelenk und die interphalangealen Gelenke (d.h. zwischen den Fingergliedern) entstehen sekundär durch einen eigens gesteuerten Mechanismus. Die molekularen Prozesse, die diese Entwicklung steuern, sind noch weitgehend unbekannt. Es konnte jedoch gezeigt werden, dass sowohl der Wnt-Signalweg wie auch BMPs und GDFs (siehe unten) eine entscheidende Rolle spielen. Ferner scheinen Hox-Gene für die frühe Musterbildungsphase der Gelenkdetermination wichtig zu sein [3].
Durch die Analyse entsprechender Tiermodelle und Erkrankungen wurde eine ganze Kaskade von molekularen Signalwegen aufgedeckt, die zum weiteren Verständnis dieses komplexen Vorgangs beitragen. So weist beispielsweise die Mausmutante short digits (Dsh) eine Veränderung der Extremitäten auf, die der humanen Erkrankung Brachydaktylie Typ A1 gleichen. Die Erkrankung ist durch eine Verkürzung bzw. Verschmelzung der mittleren Phalanx mit der proximalen Phalanx charakterisiert, sodass das mittlere interphalangeale Gelenk verloren geht und es zu einer Verkürzung der Finger kommt (Abb. 1).
. (A) Skelettpräparationen der Finger zeigen das Fehlen der mittleren Phalanx (P2). (B) Verkürzung der hinteren Extremität. (C) Skelettpräparationen (Knorpel) in einem frühen Stadium der Entwicklung (14,5 Tage) zeigen das Fehlen eines Elements sowie eine Gelenkfusion (Pfeil).")
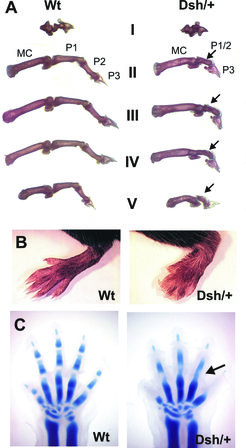
Kürzlich konnte gezeigt werden, dass der Dsh-Phänotyp durch einen Chromosomenbruch hervorgerufen wird, der das Sonic Hedgehog-Gen (Shh) von seinen regulativen Elementen trennt [4]. Dieser Chromosomenbruch, eine so genannte Inversion, führt im homozygoten Zustand zu einem weitgehenden Verlust der Shh-Expression und im heterozygoten Zustand zu einer zeitlich und örtlich falschen Expression von Shh. Der Phänotyp der homozygoten Dsh-Tiere ist daher identisch mit dem Phänotyp der Tiere, bei denen Shh homozygot inaktiviert wurde. Im Gegensatz hierzu sind die heterozygoten Shh-Tiere völlig normal.
Zeit und Raum definieren die Genwirkung
Eines der überraschendsten Ergebnisse der Analyse der Dsh-Mutante war, dass die Veränderung einerseits zu einer Deaktivierung der normalen Shh-Aktivität, andererseits jedoch zu einer ektopen Shh-Expression in den Kondensationen der Skelettanlage führte, einer Region, in der normalerweise Ihh exprimiert wird (Abb. 2).
 und die fehlende Expression von Ihh an gleicher Stelle (Pfeil) im Vergleich zu normalen Mäusen (wt) erklärt die Verkürzung eben dieser Skelettteile.")
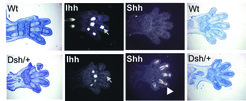
Zugleich ging die Missexpression von Shh mit einer Reduktion der endogenen Ihh-Expression einher. Eine weitere Eigenart dieser Inversion ist, dass die falsche Expression von Shh nur in den Fingern und nicht in den anderen Skelettelementen stattfindet, in denen normalerweise auch Ihh exprimiert wird. Die Inversion scheint damit zu einer neuen, chimären Regulation von Shh zu führen, die möglicherweise durch das Entfernen von so genannten Repressoren hervorgerufen wird. Wenn also ein Repressor durch die Inversion zerstört wurde, könnte möglicherweise die ektope Shh-Expression einem Teil des ursprünglichen Expressionsmusters entsprechen, was wiederum die Überlegung nahe legt, dass Shh und Ihh in einer frühen evolutionären Vergangenheit gemeinsame Expressionsmuster hatten. Dies ist nicht unwahrscheinlich, da davon ausgegangen wird, dass sich diese Gene im Laufe der evolutionären Entwicklung dupliziert und dadurch ihre spezifischen Expressionsmuster gewonnen haben.
Ein molekulares Netzwerk von Signalmolekülen reguliert die Gelenkentwicklung
Neben den Hedgehog-Molekülen sind eine Reihe von anderen Morphogenen während der Entwicklung von entscheidender Bedeutung. Zu ihnen gehören die so genannten „Bone Morphogenetic Proteins“ (BMPs) und die „Growth and Differentiation Factors“ (GDFs). BMPs sind in multiple Prozesse der Entwicklung involviert, wo sie ihre Funktion über eine Reihe von spezifischen Rezeptoren an das Zellinnere weitergeben. BMPs sind äußerst potente Morphogene, die einem extrem reglementierten Expressionsmuster folgen und durch eine ganze Familie von Inhibitoren in ihrer Funktion reguliert werden. Zu den Inhibitoren gehört u.a. Noggin, welches die BMPs wie eine Klammer umhüllt und somit eine Bindung an den Rezeptor verhindert.
Durch die Identifikation und Charakterisierung verschiedener Mutationen im GDF5, seinem Rezeptor BMPR1B und dem Inhibitor Noggin gelang es, eine komplette molekulare Regulationskaskade aufzuschlüsseln und mit bestimmten Formen von Handfehlbildungen zu koppeln [5, 6, 7]. So konnte gezeigt werden, dass Mutationen im BMP-Rezeptor 1B zu einer bestimmten Form der Fingerverkürzung (Brachydaktylie) führen [8] und dass diese Veränderung auch durch eine Mutation im GDF5 hervorgerufen werden kann. Die Mutation (L441P) verändert eine Aminosäure innerhalb des Bereiches von GDF5, der die Bindungsstelle für den BMP1B-Rezeptor darstellt [9]. Die Analyse des mutanten GDF5 durch Überexpression und Behandlung von Zellen mit rekombinanten Proteinen zeigte, dass die Mutation L441P einen nahezu kompletten Funktionsverlust aufweist, der durch eine mangelnde Bindung an seinen Rezeptor zu erklären ist.
Die Mutation R438L liegt direkt benachbart zur L441P-Mutation und somit auch innerhalb der Rezeptorbindungsdomäne. Allerdings hat diese Mutation offensichtlich einen völlig anderen Wirkungsmechanismus, da sie zum Symphalangismus führt, wie es bei Mutationen im NOGGIN-Gen beobachtet wird. Wird die Mutation R438L in Zellsystemen überexprimiert, zeigt sie eine leicht verstärkte Wirkung im Vergleich zum Wildtyp GDF5. Der eigentliche Effekt der Mutante beruht aber auf ihrer Fähigkeit, neben dem BMP-1B-Rezeptor auch den 1A-Rezeptor zu aktivieren, der normalerweise nicht durch GDF5, sondern nur durch BMP2 aktiviert werden kann (Abb. 3).
 oder Muskelzellen (unten, Myosin) differenzieren, mit verschiedenen rekombinanten GDF5. Die R438L-Mutante weist BMP2-Funktion auf.")
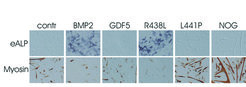
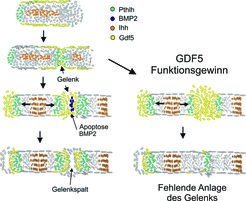
Hierdurch kommt es aller Wahrscheinlichkeit nach zu einer Bildung von zuviel Knorpel und somit zur Unterbrechung der Gelenkentstehung. So lässt sich durch eine veränderte Molekülaktivität der Phänotyp des Symphalangismus erklären (Abb. 4). Über die Veränderung von Rezeptoraffinitäten und anderen Eigenschaften eröffnen die identifizierten Varianten von GDF5 die Möglichkeit, maßgeschneiderte Moleküle zu erstellen, mit deren Hilfe die Induktion und Heilung von Knochen und Knorpel gezielt gesteuert werden können.
 und Migration. Es entsteht der zukünftige Gelenkspalt, der sich im weiteren Verlauf immer weiter ausbildet und parallel zur Gelenkkapsel entwickelt.")
Repeat-Expansionen verursachen Fehlbildungen
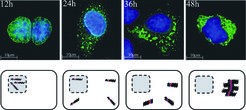
Das menschliche Genom enthält viele sich wiederholende Sequenzen, so genannte Repeats. Anscheinend sind diese Repeats besonders anfällig für Mutationen, wodurch sie Krankheiten zumeist neurologisch-degenerativer Natur verursachen. Vor kurzem wurde eine neue Form der Repeat-Expansion entdeckt, die vorwiegend in Transkriptionsfaktoren vorkommt und im Gegensatz zu den anderen bisher bekannten Repeats Sequenzen betrifft, die die Aminosäure Alanin kodieren. Im Gegensatz zu den bisher bekannten Repeats sind diese Verlängerungen kurz (Expansion von 15 Alaninen auf > 20 Alanine) und führen ausnahmslos zu angeborenen Fehlbildungen [10]. In einer aktuellen Untersuchung konnte nachgewiesen werden, dass die Repeat-Expansion zu einer falschen Faltung und damit Bildung von Aggregaten in der Zelle führt (Abb. 5) [11]. In Zellkultur kann dieser Prozess durch Medikamente, welche die Proteinfaltung beeinflussen, verbessert werden.
 und schematische Darstellung unten. Anfangs wird das mutierte Protein noch an seinen Bestimmungsort in den Zellkern transportiert, dann bilden sich im Zytoplasma kleine Aggregate, die im Verlauf zunehmend wachsen und schließlich fast die ganze Zelle ausfüllen.")
Von der Maus zum Mensch und zurück
Ziel unserer Untersuchungen ist es, menschliche Erkrankungen, insbesondere angeborene Fehlbildungen des Skeletts, besser zu verstehen, um diesen Zugewinn an Wissen zum Wohle von Patienten einsetzen zu können. Durch die bestehende Kooperation mit der Charité und die organisatorische Einheit der Forschergruppe mit dem Institut für Medizinische Genetik an der Charité sind hierfür ideale Voraussetzungen geschaffen. Die klinische Erforschung seltener genetischer Krankheitsbilder kann zusammen mit molekularbiologisch-genetischen Funktionsanalysen erfolgen und kommt direkt den betroffenen Patienten zu Gute. Einen wesentlichen Bestandteil bilden dabei neue diagnostische Möglichkeiten, mit deren Hilfe auch seltene Erkrankungen eindeutig zugeordnet werden können. Die molekularbiologisch-genetische Diagnostik von Skelettfehlbildungen wurde massiv erweitert, wodurch auch die Zahl der Konsultationen und Zusendungen stark gestiegen ist. Durch die Etablierung des Instituts als nationales und internationales Referenzzentrum ist es gelungen, eine Reihe von Erkrankungen neu zu beschreiben und molekular aufzuklären [12]. Ein Durchbruch in der Diagnostik von Deletionen gelang durch die Kooperation mit der Abteilung Molekulare Humangenetik (H.-Hilger Ropers) am Max-Planck-Institut für molekulare Genetik, wodurch die Microarray-basierte komparative genomische Hybridisierung in die Klinik eingeführt werden konnte. Die Ursachen klinisch-genetischer Variabilität werden in mehreren Projekten im Rahmen des Sonderforschungsbereiches (SFB 577) untersucht.
Regeneration des Skeletts
Ein weiteres wichtiges Arbeitsgebiet besteht in der Erforschung der Regeneration von Knochen und Knorpel und der Anwendung dieses Wissens im medizinischen Bereich. Im Gegensatz zu vielen anderen Organen besitzt das Skelett die besondere Eigenschaft, sich komplett regenerieren zu können. So wird zum Beispiel bei einem Knochenbruch neues Knochengewebe, der sogenannte Kallus, um den Bruch herum gebildet. Im weiteren Verlauf wird der Kallus zu Knochen umgebaut und verschmilzt mit den Knochenenden des Bruches, so dass es zu einer Komplettheilung kommt. Diese Regeneration beruht auf der Aktivierung von Stammzellen aus dem Knochen bzw. der näheren Umgebung der Knochenhaut, der Differenzierung der Stammzellen in chondrozytäre und osteoblastäre Vorläuferzellen und schließlich einem geordneten Auf- und Abbau von Knochenmatrix. Um die komplexe Regulation dieses Systems zu verstehen, ist die Identifikation von Schlüsselmolekülen notwendig, die zentrale Differenzierungsschritte steuern. Dies wird erreicht durch die komplette Erfassung aller während dieses Prozesses angeschalteten Gene und die nachfolgende systematische Analyse durch bioinformatische und zellbiologische Methoden. In Zusammenarbeit mit den Abteilungen Bioinformatik (Martin Vingron) und Analyse des Vertebratengenoms (Hans Lehrach) am MPI für molekulare Genetik ist es uns gelungen, Genbanken vom Kallusgewebe des Schafes anzulegen, zu sequenzieren und zu analysieren. Diese Daten bilden den Grundstock für weitere funktionelle Analysen, die wichtige Mechanismen der Genregulation und Genfunktion während des Heilungsprozesses aufdecken werden.