Die Kopplung zellulärer und mutagener Prozesse bei der Evolution von Säugetieren
Forschungsbericht (importiert) 2008 - Max-Planck-Institut für molekulare Genetik
Einführung
Mithilfe modernster Sequenziermethoden wurde in den letzten Jahren das Erbgut (Genom) von mehr als 30 Säugetieren charakterisiert. Die Vielzahl an bekannten Genomen ermöglicht heute vergleichende Untersuchungen zur evolutionären Entwicklung und Dynamik der Erbanlagen. Unterschiede in kodierenden Bereichen der DNA können auf Strukturänderungen eines einzelnen Proteins oder eines regulatorischen Netzwerkes hindeuten. Solche Unterschiede können adaptiver Natur sein, also der betreffenden Spezies bei der Anpassung an sich ändernde Lebensumstände helfen, oder aber zufällig durch evolutionär neutrale Mutationen der DNA entstanden sein. Zur Unterscheidung dieser beiden Möglichkeiten ist die genaue Kenntnis der durch Zufall verursachten Änderungen von genomischen DNA-Sequenzen unumgänglich. Je nach Spezies sind hier unterschiedliche Prozesse von Bedeutung. Im Folgenden sollen mutagene Prozesse in Wirbel- und Säugetiergenomen und deren kürzlich erforschte Wechselwirkungen mit anderen zellulären Prozessen vorgestellt werden. Solche Wechselwirkungen verschiedener Prozesses sind gerade bei der Betrachtung der Evolution von Säugetierarten wichtig, da diese häufig nur kleine Populationsgrößen aufweisen und somit der Einfluss der natürlichen Selektion reduziert ist [1].
Die Prozesse der Genomevolution
Einer der häufigsten Prozesse bei der Veränderung der DNA ist der Basenaustausch an einer bestimmten Position. Solch ein Basenaustausch findet zunächst auf einem Strang der doppelsträngigen DNA statt. Üblicherweise verbinden sich innerhalb des DNA-Doppelstranges stets Cytosin (C) mit Guanin (G) und Adenin (A) mit Thymin (T). Ein Basenaustausch führt entsprechend zu einer Fehlstelle innerhalb der DNA (Abb. 1).
; diese Fehlstelle wird dann zufällig auf eine von zwei Weisen ausgebessert (Reparatur). Falls weder der Mutations- noch der Reparaturprozess die beiden Stränge der DNA unterscheiden können, ist die Rate einer Substitution von A nach C gleich derjenigen für eine Substitution von T nach G.")
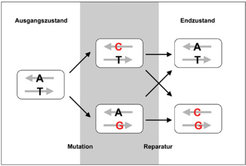
Die Zelle verfügt über Mechanismen, solche Fehlstellen zu erkennen und zu reparieren. Da diesem Reparaturmechanismus allerdings keine Information über den Zustand der DNA vor der Mutation zur Verfügung steht, wird zufällig entweder der Anfangszustand wieder hergestellt oder aber der komplementäre Strang der DNA verändert, um die richtige Basenpaarung zu gewährleisten. Die einfachsten Modelle für den Austausch von Basenpaaren gehen davon aus, dass jeder Austausch unabhängig von allen anderen Basen entlang der Sequenz stattfindet, und dass der Reparaturprozess die beiden Stränge der DNA nicht voneinander unterscheiden kann. Dies führt dazu, dass entlang eines Stranges etwa gleich viele Übergänge von C nach T wie von G nach A zu beobachten sind.
Weiterentwicklungen der Modelle für Basenaustausche
Die Fülle der heute verfügbaren Genomdaten erlaubt es, die einfachen Modelle des Basenaustausches weiterzuentwickeln. So vernachlässigen diese Modelle die Methylierung von Cytosin, die einem Guanin benachbart sind (CpGs). Die Methylierung von CpGs ist ein biochemischer Prozess, der bei der Genregulation in Wirbeltieren eine wichtige Rolle spielt. Methylierte CpGs am Beginn kodierender Regionen der DNA signalisieren, dass die entsprechenden Gene nicht abgelesen werden können. Die Methylierung eines CpGs wird auch bei der Zellteilung aufrechterhalten, sodass ein Gen durch diesen Vorgang dauerhaft ausgeschaltet werden kann. Allerdings hat die Methylierung auch einen mutagenen Einfluss auf die DNA-Sequenz: Methyliertes Cytosin kann sehr leicht zu Thymin mutieren, da hierzu lediglich eine Aminogruppe entfernt werden muss (Desamination). Infolgedessen wird CpG häufig durch TpG ausgetauscht, was dem Austausch eines CpG durch ein CpA auf dem Gegenstrang entspricht.
Im Gegensatz zu dem oben vorgestellten Modell ist der CpG-Methylierungs- und Desaminationsprozess abhängig von den jeweils benachbarten Basenpaaren, da die Cytosinmethylierung nur bei CpGs stattfindet. Bei der mathematischen Formulierung eines erweiterten Modells muss demnach auch die Identität von benachbarten Basen mit einbezogen werden. Das erweiterte Modell konnte überzeugend zeigen, dass der nachbarabhängige CpG-Methylierungs- und Desaminationsprozess den wichtigsten Vorgang bei der Veränderung unseres Erbgutes darstellt [2]. Seine Rate ist etwa 20-mal größer als die Rate der anderen Prozesse, bei denen eine Base durch eine andere ausgetauscht wird. Dies hat im Laufe der Evolution zu einer statistischen Unterrepräsentation von CpGs gegenüber TpGs und CpAs im menschlichen Genom geführt. Die Verschiebung der Zweibasen-Verteilung ist somit ein Nebeneffekt eines zellulären Prozesses, der der Regulation von Genen dient. Diese Kopplung zweier Prozesse führt zu einer Verschiebung in den Mutationsraten der unterschiedlichen Mutationsprozesse. Wenn auch diese Veränderungen für sich betrachtet klein sind, so können sie doch über die Dauer der Evolution eine deutliche Signatur in den genomischen DNA-Sequenzen hinterlassen.
Dies gilt auch für andere Prozesse, deren Kopplung an mutagene Prozesse noch schwächer ist als bei der CpG-Methylierung. So konnte vor kurzem gezeigt werden, dass auch die Transkription der DNA in RNA einen Einfluss auf die in kodierenden Bereichen auftretenden Mutationen hat. Während der Transkription muss der DNA-Doppelstrang getrennt werden. Nur einer der beiden Stränge wird durch die RNA-Polymerase abgelesen und in eine komplementäre RNA-Sequenz übersetzt. Während dieses Vorganges ist der DNA-Gegenstrang ungebunden und deshalb anfälliger für Mutationen. In transkribierten Bereichen ist daher die Symmetrie gebrochen, nach der die beiden Stränge der DNA gleich beeinflusst werden (vgl. Abb. 1). Die Brechung dieser Symmetrie kann in einigen Fällen zu einer Veränderung der Mutationsraten führen. Eine solche Veränderung konnte beim Vergleich von transkribierten Sequenzen des menschlichen Genoms mit denen des Schimpansen und des Rhesusaffen nachgewiesen werden [3]. Abbildung 2 zeigt die Profile der 12 verschiedenen Mutationsprozesse sowie die Raten der CpG-Methylierung und Desamination. Während in nicht-transkribierten Regionen vor Genen die Raten von komplementären Prozessen in etwa gleich sind, ist dies in den transkribierten Bereichen nicht mehr der Fall. Dies beweist die Kopplung mutagener Prozesse an die Transkription.

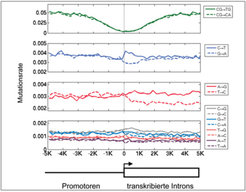
Entlang der DNA-Sequenz können drei unterschiedliche Muster der Symmetriebrechung beobachtet werden. In einem Bereich von 1 kbp (kilo Basenpaaren) vor bis etwa 1 kbp nach dem Transkriptionsstart ist der CpG-Methylierungs- und Desaminationsprozess unterdrückt. Dagegen zeigen die nachbarunabhängigen Prozesse erst unmittelbar nach dem Transkriptionsstart die erwartete Symmetriebrechung. Zum Beispiel beobachtet man entlang des kompletten Transkripts mehr Übergänge von A nach G als von T nach C (vgl. Abb. 2). Weiterhin ist auffällig, dass Übergänge von C nach T gegenüber Übergängen von G nach A nur lokal etwa 1500 bp hinter dem Transkriptionsstart asymmetrisch auftreten. Letztere lokalisierte Asymmetrie muss deshalb durch einen weiteren Mechanismus verursacht werden.
Die vorgelegten Analysen liefern wichtige Informationen über komplexe biochemische Vorgänge, deren Einfluss auf die Evolution durch Experimente nur schwer zur quantifizieren wäre. Dies gilt auch für weitere Prozesse, wie zum Beispiel die Rekombination. Bei der Rekombination kommt es oft zur Ausbildung von Heteroduplex-Strukturen der DNA, die bei Vorhandensein einer Mutation auf einem der beiden Chromatiden präferenziell zu einem GC-Basenpaar verändert werden. Auch hier konnte der Einfluss der Rekombination auf Veränderungen der Basenkomposition der DNA durch eine vergleichende Genomanalyse quantifiziert werden [4].
Einfügungen und Löschungen innerhalb der DNA
Auch Prozesse, die die Länge der genomischen Sequenz verlängern oder verkürzen, spielen eine wichtige Rolle für die Veränderung von Genomen. Solche Vorgänge finden in der Regel viel seltener als Basenaustausche statt. In der Evolution der Wirbeltiere kann am häufigsten die Einfügung weiterer Kopien von repetitiven Elementen beobachtet werden. Diese Elemente machen heute etwa 46% des menschlichen Genoms aus [5]. Daneben werden aber auch kleinere Sequenzstücke neu in Genome eingefügt. Durch vergleichende Genomanalyse konnte gezeigt werden, dass es sich dabei in der Regel nicht um zufällige Sequenzen handelt, sondern dass sehr oft ein Teil der DNA verdoppelt und gleich neben dem originalen Abschnitt wieder eingesetzt wurde. Diese Signatur weist deutlich auf einen Prozess hin, der bei der Reparatur von Doppelstrangbrüchen der DNA zum Tragen kommt [6]. Im Gegensatz zum Basenaustausch bietet die Verdopplung von DNA-Segmenten die Möglichkeit, größere Sprünge bei der Evolution neuer funktionaler Elemente zu machen. So kann zum Beispiel ein für ein Protein kodierender Bereich verdoppelt werden; das Original behält dann seine Funktion innerhalb der Zelle, während die Kopie durch kleine Veränderungen eine neue Funktion entwickeln kann.
Ausblick
Für die mathematische Beschreibung von Basenaustauschen existieren mehrere Modelle. Wichtige Weiterentwicklungen auf diesem Gebiet stellen die Einbeziehung von Nachbarabhängigkeiten entlang der DNA sowie die Beachtung anderer zellulärer Prozesse dar. Die vergleichende Analyse von Genomen konnte so weit verallgemeinert werden, dass auch nicht reversible Modelle des Basenaustauschs verwendet werden können [7]. Erstmalig konnte gezeigt werden, dass sich genomische Sequenzen entgegen einer weit verbreiteten Annahme nicht in einem stationären Zustand befinden, sondern sehr wohl auch ihre Basenkomposition ändern.
Der in der nächsten Zeit zu erwartende Anstieg an genomischen Sequenzdaten stellt eine gewaltige Herausforderung an die Analyse dieser Daten dar. Es wird vor allem darauf ankommen, diese Daten mit geeigneten Mitteln zu analysieren, um so wissenschaftliche Fragestellungen bearbeiten zu können. Dabei wird der mathematischen Modellierung eine Schlüsselrolle zufallen. Nur der Vergleich genomischer Sequenzen verschiedener Lebewesen ermöglicht es, die Geschichte der Evolution weiter aufzudecken. Daneben wird das Wissen um biologische Prozesse und deren Entwicklung weiter wachsen, sodass wir in Zukunft ein umfassenderes Bild der komplexen Abläufe in einer Zelle bekommen werden.