Gib mir fünf! Oder sechs? Oder sieben?
Forschungsbericht (importiert) 2009 - Max-Planck-Institut für molekulare Genetik
Hox-Gene während der embryonalen Entwicklung

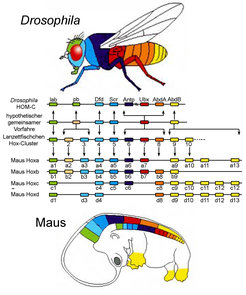
Im Verlauf der embryonalen Entwicklung müssen verschiedene Achsen im sich entwickelnden Embryo angelegt werden, so zum Beispiel die primäre Körperachse vom Kopf zum Schwanz oder körpernah bis körperfern in den Extremitäten. Die Familie der Hox-Gene, eine hoch konservierte Familie von Transkriptionsfaktoren, spielt dabei eine wichtige Rolle [1]. Charakteristisch für diese Genfamilie, die wiederum andere Faktoren steuert, ist das Vorhandensein der namensgebenden Homöobox, einer konservierten Sequenz, die ein spezifisches DNA-Bindungsmotiv kodiert und so die Regulation anderer Gene ermöglicht.
Hox-Gene sind in Clustern organisiert und haben sich vermutlich, ausgehend von einem gemeinsamen Vorfahren, im Zuge der Evolution durch Verdopplungsereignisse entwickelt. Die Fruchtfliege besitzt einen Cluster, der aus dem Antennapedia und Bithorax Komplex besteht. Wirbeltiere dagegen weisen vier Cluster - HoxA, HoxB, HoxC und HoxD - auf, die insgesamt 39 Gene umfassen. Während der embryonalen Entwicklung folgen die Hox-Gene dem Prinzip der räumlichen und zeitlichen Kolinearität. Dies bedeutet, dass Hox-Gene, die exakt kontrolliert werden, in genau festgelegter Weise nacheinander angeschaltet werden. Dabei sind nicht nur der Zeitpunkt, sondern auch die Position auf der Körperachse, an der das jeweilige Gen aktiviert wird, genauestens definiert (Abb. 1).
Hox-Gene und Synpolydaktylie

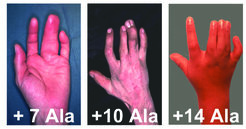
In der Medizin sind eine Reihe humaner Fehlbildungen bekannt, die auf Mutationen in den Hox-Genen zurückzuführen sind. Eine dieser Fehlbildungen ist die Synpolydaktylie (SPD), eine erblich bedingte Skelettfehlbildung der distalen Extremitäten. Der Begriff Synpolydaktylie stammt aus dem Griechischen und beinhaltet eine Vielfingrigkeit (poly – viel; dactylos – der Finger), bei der zusätzlich einzelne Elemente fusioniert sind (syn – zusammen). Die Fehlbildung an Händen und Füßen wird von einer dominanten Mutation im Hoxd13-Gen ausgelöst. Bei dieser Mutation handelt es sich um die Verlängerung einer Alanin-Expansion innerhalb der kodierenden Gensequenz: Normalerweise beinhaltet diese Expansion, das heißt, mehrfache Wiederholung der gleichen Aminosäure, 15 Alanine. Im Krankheitsfall jedoch ist die Reihe um mindestens sieben Alanine verlängert (Abb. 2). Besonders interessant dabei ist, dass der Phänotyp der Krankheit umso schwerwiegender ausfällt, je mehr Alanine hinzugefügt sind. Die Erkrankung wird dominant vererbt, es genügt also bereits eine Kopie des mutierten Gens (von Mutter oder Vater), um das Krankheitsbild zu entwickeln. Sind beide Kopien des Hoxd13-Gens verändert (homozygot), wirkt sich die Mutation noch schwerwiegender aus.
Denn sie wissen nicht, was sie tun
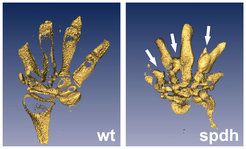
Wenn Mäusen das Hoxd13-Gen fehlt, entwickeln sie keine Synpolydaktylie. Das bei der Erkrankung entstehende veränderte, verlängerte Genprodukt (Protein) muss also einen negativen Effekt auf andere Faktoren ausüben. Um die dem Krankheitsbild zugrunde liegenden Mechanismen im Verlauf der Embryogenese aufzuklären, wurde das Mausmodell spdh untersucht (Abb. 3). Spdh-Mäuse besitzen eine Mutation, die homolog zu der menschlichen Mutation bei Synpolydaktylie ist und in Mäusen den gleichen Phänotyp auslöst. Zwei verschiedene Mechanismen wurden gefunden, die dafür verantwortlich sind, dass zu viele Finger entstehen, die miteinander fusionieren.
Zum einen aktiviert Hoxd13 das Enzym Raldh2, das in der Extremitätenknospe Retinsäure aus Vitamin A produziert. Retinsäure ist ein wichtiger Signalgeber während der Musterbildung und verhindert, dass sich die Zellen in den Fingerzwischenräumen zu Knorpelzellen spezialisieren. Diese Spezialisierung ist der erste Schritt in der Entwicklung von embryonalen Vorläuferzellen hin zum späteren Fingerknochen. Die mutierte Proteinvariante des Hoxd13-Gens jedoch ist nicht in der Lage, Raldh2 ausreichend zu aktivieren. In Folge entsteht weniger Retinsäure und die Zellen in den Fingerzwischenräumen erhalten falsche Signale. Sie entwickeln sich ebenfalls zu Knorpelzellen, aus denen im Laufe der Embryonalentwicklung überzählige Finger entstehen. Zusätzlich fusionieren Finger miteinander, weil die Grenzen zwischen Finger und Fingerzwischenraum nicht korrekt gezogen werden konnten. Behandelt man schwangere spdh-Muttertiere mit Retinsäure und gleicht damit den entstandenen Mangel in den Embryonen aus, werden Nachkommen geboren, die zwar den genetischen Defekt tragen, aber dennoch mit fünf Fingern zur Welt kommen. Zum anderen hat Hoxd13 selbst einen knorpelhemmenden Effekt. Dieser geht bei der verlängerten Variante verloren, wodurch die Spezialisierung der Vorläuferzellen hin zur Knorpelzelle weiter verstärkt wird [3].
. Im gesunden Wildtyp (wt) sind deutlich die knorpeligen Fingeranlagen erkennbar. Im Modell der spdh-Mutante erkennt man zusätzlichen Knorpel in den Fingerzwischenräumen, der teilweise mit den Fingeranlagen fusioniert ist (Pfeile).")
Durch die Untersuchung der molekularen Mechanismen der Synpolydaktylie konnten zwei grundsätzliche Sachverhalte aufgeklärt werden. Bisher gingen Wissenschaftler davon aus, dass zusätzliche Finger durch die Verdoppelung bereits vorhandener Fingeranlagen entstehen, es sich also um Duplikationen handelt. Für bestimmte Skelettfehlbildungen kann dieser Mechanismus auch nachgewiesen werden. Bei dem hier beschriebenen Krankheitsbild handelt es sich jedoch um einen Differenzierungsdefekt. Zellen in den Fingerzwischenräumen bekommen falsche Signale und beginnen mit einer unkontrollierten Spezialisierung. Außerdem war bisher nur bekannt, dass Hox-Gene von entscheidender Bedeutung für die Achsenanlage und Musterbildung im Embryo sind. Die vorliegenden Ergebnisse zeigen jedoch, dass sie auch bei späteren Prozessen der embryonalen Entwicklung eine wichtige Rolle spielen und ihre Bedeutung bisher eher unterschätzt wurde. Diese Bedeutung wird derzeit weiter untersucht.