Neurodegenerative Erkrankungen - von der Bäckerhefe zu neurodegenerativen Prozessen
Forschungsbericht (importiert) 2010 - Max-Planck-Institut für molekulare Genetik
Einleitung
Neurodegenerative Erkrankungen sind durch einen fortschreitenden Verlust von Nervenzellen gekennzeichnet und zählen heutzutage neben Krebserkrankungen zu den häufigsten Todesursachen. Da die Lebenserwartung in den westlichen Nationen eine stetige Zunahme verzeichnet, stellen die spätmanifesten neurodegenerativen Erkrankungen, wie zum Beispiel die Alzheimer- beziehungsweise Parkinson-Krankheit oder auch die Familie der Polyglutamin-Erkrankungen, ein in Zukunft nicht zu unterschätzendes Gesundheitsproblem dar. Weltweit leiden Millionen von Menschen unter diesen Erkrankungen oder besitzen das Risiko, zu erkranken, sodass ein weiterer erheblicher Anstieg der wirtschaftlichen und sozialen Belastungen in den nächsten Jahrzehnten zu erwarten ist. Effektive therapeutische Strategien zur Prävention oder Heilung dieser verheerenden Erkrankungen stehen bis heute nicht zur Verfügung.
Die Polyglutamin-Erkrankung Spinozerebellare Ataxie Typ 2
Der Forschungsschwerpunkt der Arbeitsgruppe liegt auf der Identifizierung von molekularen Mechanismen, die zur Pathogenese der Polyglutamin-Erkrankung Spinozerebellare Ataxie Typ 2 (SCA2) beitragen können. Weltweit gesehen ist diese Erkrankung eine selten auftretende progressive neurodegenerative Erkrankung, gehört aber innerhalb der Polyglutamin-Erkrankungen zu den drei häufigsten aller autosomal dominant vererbbaren Spinozerebellaren Ataxien und repräsentiert auch in Deutschland mit 15% die dritthäufigste Form. Im Verlauf von SCA2 werden klinische Manifestationen wie zum Beispiel bedingte Gang- und Standataxie, Dysarthrie, Sakkadenverlangsamung und Tremor beobachtet, wobei sich diese klinischen Profile im Krankheitsverlauf verstärken. Ursächlich für diese Symptomatik sind eine erhebliche Degeneration von Purkinje-Zellen im Kleinhirn. Des Weiteren wird ein neuronaler Verlust der Substantia nigra beobachtet, der sich in Parkinson-ähnlicher Rigidität und Bradykinesia manifestiert [1]. Auf molekularer Ebene ist SCA2 durch eine Verlängerung von CAG-Trinukleotidwiederholungen in der kodierenden Region des sogenannten SCA2-Gens charakterisiert. Zudem besteht eine inverse Korrelation zwischen der Schwere der klinischen Symptomatik beziehungsweise dem Beginn der Erkrankung und der Verlängerung des Trinukleotidbereichs, das heißt, je mehr Trinukleotidwiederholungen vorhanden sind, desto früher erkranken die betroffenen Personen und desto ungünstiger ist auch die Prognose [1]. Auf Proteinebene führt eine Verlängerung von CAG-Trinukleotidwiederholungen im SCA2-Gen zu einem verlängerten Glutaminbereich in der N-terminalen Region des SCA2-Genprodukts, Ataxin-2. Ein Hauptmerkmal vieler neurodegenerativer Erkrankungen ist die Bildung von abnormalen Proteinablagerungen oder sogenannten Proteinaggregaten in den jeweiligen betroffenen Gehirnregionen. Interessanterweise scheint bei SCA2 die neuronale Degeneration eher auf einer Zunahme der intrazellulären Konzentration von Ataxin-2 als auf der Bildung von abnormalen Proteinablagerungen zu beruhen [2-3].
Trotz intensiver Untersuchungen sind sowohl die biologische Funktion der meisten Polyglutamin-Proteine als auch die zellulären Mechanismen, welche zur Pathogenese beitragen, weitestgehend unbekannt oder nur unvollständig verstanden. Dies gilt auch für Ataxin-2. Einblicke in die zelluläre Funktion von neurodegenerativen Proteinen sind jedoch für die Identifizierung potenzieller Pathomechanismen von enormer Bedeutung. Daher wurden in den letzten Jahren verschiedene Modellsysteme entwickelt und zur Identifizierung von zellulären Faktoren, welche Aggregation, zelluläre Dysfunktion und Zelltod beeinflussen, eingesetzt.
Bäckerhefe als Modellsystem
Die Bäckerhefe Saccharomyces cerevisiae, die den am besten charakterisierten, einzelligen Eukaryonten darstellt, stellt ein relevantes und genetisch sehr gut zu manipulierendes Modellsystem für neurodegenerative Erkrankungen dar. Von Bedeutung ist, dass generelle zelluläre Mechanismen wie Zellteilung, Proteinfaltung, zelluläre Stressantworten, Alterung, Metabolismus, Zelltod etc. bei Hefe und höheren Eukaryonten - also auch dem Menschen - sehr konserviert sind. Umfassende genetische Analysen in Hefe führten bereits zur Identifizierung von Genen, die die Toxizität und/oder Aggregation von humanen neurodegenerativen Proteinen modulieren [4]. Zudem existieren für eine Vielzahl von humanen Genen Homologe in der Hefe; darunter befinden sich auch Gene, die mit humanen Erkrankungen in Verbindung gebracht worden sind.

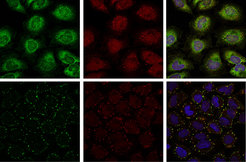
Auch für das Protein Ataxin-2 wurde ein Homolog, Pbp1p, in der Bäckerhefe beschrieben. Die Proteinarchitektur des Hefe-Homologs Pbp1p und von Ataxin-2 sind sehr ähnlich, was auf potenziell konservierte zelluläre Funktionen in der Hefe und in humanen Zellen hinweisen könnte. Eine vergleichende Interaktomanalyse des entsprechenden Hefe-Homologs Pbp1p ergab, dass die untersuchten Protein-Protein-Wechselwirkungen des Hefeinteraktoms direkt auf das humane System übertragen werden konnten. Von den identifizierten Protein-Protein-Interaktionen ausgehend konnte die Arbeitsgruppe demonstrieren, dass Ataxin-2 eine Komponente der so genannten stress granules darstellt und die Zusammensetzung dieser zellulären Strukturen beeinflusst (Abb. 1; [5, 6]). Stress granules sind zytoplasmatische Strukturen, die innerhalb kurzer Zeit im Zytoplasma von Säugerzellen als Antwort auf verschiedene Stressbedingungen wie zum Beispiel Hitzestress, oxidativen und osmotischen Stress etc. entstehen [7]. Obwohl stress granules einen grundlegenden adaptiven Schutzmechanismus der Zelle darstellen, ist die biologische Verknüpfung mit anderen zellulären Prozessen, zum Beispiel zellulären Signalwegen, noch nicht ausreichend verstanden. Anzumerken ist zudem, dass sowohl die zelluläre Zusammensetzung von stress granules als auch deren Bildung und Bedeutung für humane Erkrankungen noch nicht vollständig geklärt sind und in Zukunft weiterer Analysen bedürfen.
Erste Ergebnisse der Arbeitsgruppe weisen darauf hin, dass Veränderungen im zellulären RNA-Metabolismus potenziell zur Pathogenese von SCA2 beitragen könnten, da beispielsweise die Überexpression von Ataxin-2 die Bildung von processing bodies, die mit RNA-Degradation in Verbindung gebracht worden sind, beeinträchtigt [6]. Interessanterweise scheinen Beeinträchtigungen im zellulären RNA-Metabolismus auch bei anderen neurodegenerativen Erkrankungen von Bedeutung zu sein.
Überlappende Mechanismen in neurodegenerativen Erkrankungen?
Mit Hilfe transgener Drosophila-Modelle für die Spinozerebellare Ataxie Typ 1 bzw. 3 sowie für die amyotrophe Lateralsklerose konnte gezeigt werden, dass Ataxin-2 Einfluss auf die jeweils beobachtete Toxizität nimmt [8-10]. Somit wird eine Beteiligung von Ataxin-2 an zellulären Pathomechanismen anderer neurodegenerativer Erkrankungen diskutiert.
Weitere Analysen werden helfen, in der Zukunft diese zellulären Zusammenhänge tiefgreifend zu entschlüsseln.